algoritmo de Smith-Waterman es una reconocida estrategia para realizar alineamiento local de secuencias biológicas (ADN, ARN o proteínas); es decir que determina regiones similares entre un par de secuencias.
El algoritmo SW fue propuesto por Temple Smith y Michael Waterman en 1981.1 Está basado en el uso de algoritmos de programación dinámica, de tal forma que tiene la deseable propiedad de garantizar que el alineamiento local encontrado es óptimo con respecto a un determinado sistema de puntajes que se use (tales como matrices de substitución).
Las alternativas básicas para realizar el alineamiento de un par de secuencias son: el alineamiento local y el alineamiento global. Los alineamientos globales pretenden alinear cada símbolo (o residuo) en cada secuencia. Esta estrategia es especialmente útil cuando las secuencias a alinear son altamente similares y aproximadamente del mismo tamaño. En contraste, los alineamientos locales son más útiles cuando las secuencias a alinear poseen grandes diferencias, pero se sospecha que existen regiones de similitud.
alineamiento de secuencias en bioinformática es una forma de representar y comparar dos o más secuencias o cadenas de ADN, ARN, o estructuras primarias proteicas para resaltar sus zonas de similitud, que podrían indicar relaciones funcionales o evolutivas entre los genes o proteínas consultados. Las secuencias alineadas se escriben con las letras (representando aminoácidos o nucleótidos) en filas de una matriz en las que, si es necesario, se insertan espacios para que las zonas con idéntica o similar estructura se alineen.

Si dos secuencias en un alineamiento comparten un ancestro común, las no coincidencias pueden interpretarse como mutaciones puntuales (sustituciones), y los huecos como indels (mutaciones de inserción o deleción) introducidas en uno o ambos linajes en el tiempo que transcurrió desde que divergieron. En el alineamiento de secuencias proteicas, el grado de similitud entre los aminoácidos que ocupan una posición concreta en la secuencia puede interpretarse como una medida aproximada de conservación en una región particular, o secuencia motivo, entre linajes. La ausencia de sustituciones, o la presencia de sustituciones muy conservadas (la sustitución de aminoácidos cuya cadena lateral tiene propiedades químicas similares) en una región particular de la secuencia indica que esta zona tiene importancia estructural o funcional. Aunque las bases nucleotídicas del ADN y ARN son más similares entre sí que con los aminoácidos, la conservación del emparejado de bases podría indicar papeles funcionales o estructurales similares. El alineamiento de secuencias puede utilizarse con secuencias no biológicas, como en la identificación de similitudes en series de letras y palabras del lenguaje humano o en análisis de datos financieros.
Secuencias muy cortas o muy similares pueden alinearse manualmente. Aun así, los problemas más interesantes necesitan alinear secuencias largas, muy variables y extremadamente numerosas que no pueden ser alineadas por humanos. El conocimiento humano se aplica principalmente en la construcción de algoritmos que produzcan alineamientos de alta calidad, y ocasionalmente ajustando el resultado final para representar patrones que son difíciles de introducir en algoritmos (especialmente en el caso de secuencias de nucleótidos). Las aproximaciones computacionales al alineamiento de secuencias se dividen en dos categorías: alineamiento global y alineamiento local. Calcular un alineamiento global es una forma de optimización global que "fuerza" al alineamiento a ocupar la longitud total de todas las secuencias introducidas (secuencias problema). Comparativamente, los alineamientos locales identifican regiones similares dentro de largas secuencias que normalmente son muy divergentes entre sí. A menudo se prefieren los alineamientos locales, pero pueden ser más difíciles de calcular porque se añade el desafío de identificar las regiones de mayor similitud. Se aplican gran variedad de algoritmos computacionales al problema de alineamiento de secuencias, como métodos lentos, pero de optimización, como la programación dinámica, y métodos heurísticos o probabilísticos eficientes, pero no exhautivos, diseñados para búsqueda a gran escala en bases de datos.
Representaciones[editar]
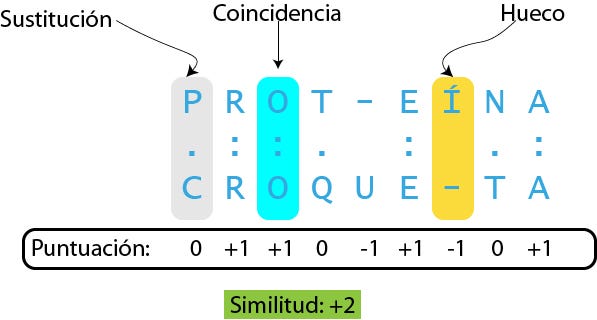
Los alineamientos se representan normalmente con un formato gráfico y de texto. En casi todas las representaciones de alineamientos, las secuencias se escriben en filas de forma que los residuos alineados aparecen en columnas sucesivas. En los formatos de texto, las columnas alineadas contienen caracteres idénticos o similares, estos últimos indicados con sistema de símbolos de conservados. En la imagen superior se utiliza el asterisco para mostrar identidad entre dos columnas. Otros símbolos menos comunes son la coma para sustituciones conservativas y el punto para sustituciones semiconservativas. Muchos programas de visualización de secuencias utilizan también esquemas coloreados para mostrar información de las propiedades de los elementos secuencia individuales; en secuencias de ADN y ARN significa asignar a cada base su propio color. En alineamientos de proteínas, como el de la imagen superior, los colores se utilizan para indicar propiedades de los aminoácidos para ayudar en la caracterización de conservación o en una sustitución aminoacídica dada. Cuando se introducen múltiples secuencias la última fila de cada columna suele representar la secuencia consenso determinada por el alineamiento. También suele representarse la secuencia consenso en un formato gráfico bajo un logo de secuencias, en el que el tamaño de la letra de cada nucleótido o aminoácido es proporcional a su grado de conservación.1
Los alineamientos de secuencias pueden almacenarse en una amplia variedad de formatos de archivo de texto, muchos de los cuales han sido desarrollados a la vez que un programa o implementación de alineamiento. La mayoría de las herramientas web permiten varios formatos de entrada y salida, como el formato FASTA y GenBank. La utilización de herramientas específicas en cada laboratorio de investigación puede complicarse por la baja compatibilidad. Existen programas de conversión genérica en SEQRET (EMBOSS), o en DNA Baser.
Alineamientos locales y globales[editar]

Los alineamientos globales, que intentan alinear cada residuo de cada secuencia, son más útiles cuando las secuencias problema iniciales son similares y aproximadamente del mismo tamaño (no quiere decir que los alineamientos globales no puedan terminar en huecos). Una estrategia general de alineamiento global es el algoritmo Needleman-Wunsch basado en programación dinámica. Los alineamientos locales son más útiles para secuencias diferenciadas en las que se sospecha que existen regiones muy similares o motivos de secuencias similares dentro de un contexto mayor. El algoritmo Smith-Waterman es un método general de alineamiento local basado en programación dinámica. Con secuencias suficientemente similares, no existe diferencia entre alineamientos globales y locales.
Los métodos híbridos, conocidos como semiglobales o métodos "glocales" intentan encontrar el mejor alineamiento posible que incluya el inicio y el final de una u otra secuencia. Puede ser especialmente útil cuando la parte "corriente arriba" de una secuencia se solapa con la parte "corriente abajo" de la otra. En este caso, ni el alineamiento global ni el local son completamente adecuados: un alineamiento global intentará forzar a la alineación a extenderse más allá de la región de solapamiento, mientras que el alineamiento local no cubrirá totalmente la región solapada.2 3
Alineamiento de pares[editar]
Los métodos de alineamiento de pares, o emparejamientos, se utilizan para encontrar la mejor coincidencia en bloque (local) o alineamiento global de dos secuencias. Los alineamientos de pares sólo pueden utilizarse con dos secuencias a la vez, pero son eficientes de calcular, y son utilizados a menudo en métodos que no requieren precisión extrema, como la búsqueda en bases de datos de secuencias con alta homología de secuencia con respecto a una petición. Los tres métodos principales de generar alineamientos de pares son los de matriz de puntos, los de programación dinámica y los de búsqueda de palabra,4 aunque la mayoría de métodos de alineación múltiple de secuencias pueden funcionar con sólo dos secuencias. Aunque cada método tiene sus propios puntos fuertes y débiles, todos ellos tienen problemas para alinear secuencias repetitivas con bajo contenido en información, especialmente cuando el número de repeticiones puede ser diferente en las dos secuencias que se alinean. Una manera de cuantificar la utilidad de un alineamiento de pares determinado es la "máxima coincidencia individual", o la mayor subsecuencia que se da en ambas secuencias en estudio. En general, cuanto mayor sea tal subsecuencia, más cercana será su relación.
Métodos de matriz de puntos[editar]

El enfoque de matriz de puntos (o matrix-dot), que implícitamente produce una familia de alineamientos para regiones individuales de la secuencia, es cualitativo y simple, a pesar de que consume mucho tiempo para análisis a gran escala. Es fácil identificar visualmente determinadas características de la secuencia (tales como inserciones, borrados, repeticiones, o repeticiones invertidas) en una gráfica de matriz de puntos. Para construir una gráfica de matriz de puntos, las dos secuencias se colocan a lo largo de la fila superior y de la columna que está más a la izquierda de la matriz de dos dimensiones y se coloca un punto en cualquier lugar en el que los caracteres en las columnas correspondientes coincidan (esta es una típica gráfica recurrente). Algunas implementaciones varían el tamaño o la intensidad de los puntos en función del grado de similitud de los dos caracteres, para dar cabida a las sustituciones conservadas. La gráfica de puntos de secuencias muy estrechamente relacionadas aparece como una única línea a lo largo de la diagonal principal de la matriz .
Las gráficas de puntos también pueden utilizarse para evaluar repetitividad en una sola secuencia: una secuencia se gráfica contra sí misma, y las regiones que comparten similitudes significativas aparecerán como líneas fuera de la diagonal principal. Este efecto puede ocurrir cuando una proteína consta de múltiples dominios estructurales similares.
Programación dinámica[editar]
La técnica de programación dinámica puede aplicarse para producir alineamientos globales mediante el algoritmo de Needleman-Wunsch, así como alineamientos locales mediante el algoritmo de Smith-Waterman. En una utilización habitual, en los alineamientos de proteínas se utiliza una matriz de sustitución para asignar puntuaciones a las coincidencias y a las diferencias entre aminoácidos, y una penalización por gap (literalmente hueco, aunque en buena parte de la literatura en castellano se utiliza el vocablo inglés) al hacer coincidir un aminoácido de una secuencia con un hueco en otra. En alineamientos de ADN y ARN puede usarse una matriz de puntuaciones, pero en la práctica, a menudo, se asigna simplemente una puntuación positiva a las coincidencias, una negativa a las diferencias, y otra penalización negativa a los gaps. (En la programación dinámica estándar la puntuación de la posición de cada aminoácido es independiente de la identidad de sus vecinos, por lo que los efectos del apilamiento de bases no se toman en cuenta. Sin embargo, es posible hacerlo modificando el algoritmo.)
La programación dinámica puede ser útil en el alineamiento de nucleótidos con secuencias de proteínas, una tarea complicada por la necesidad de tomar en consideración las mutaciones con cambio (inserciones o borrados, normalmente). La búsqueda del marco abierto de lectura proporciona una serie de alineamientos de pares locales o globales entre una secuencia de nucleótidos a investigar (secuencia problema) y un conjunto de búsqueda de secuencias de proteínas, o viceversa. A pesar de que el método es muy lento, su habilidad para evaluar la compensación de los marcos de lectura para un número arbitrario de nucleótidos lo convierte en útil para secuencias que contengan un buen número de indels, los cuales pueden ser muy difíciles de alinear con métodos heurísticos más eficientes. En la práctica, el método requiere una buena cantidad de potencia de cómputo, o un sistema cuya arquitectura esté especializada en programación dinámica. Las suites BLAST y EMBOSS proporcionan herramientas básicas para crear alineamientos adaptados (aunque alguno de estos enfoques saca ventaja de los efectos laterales de la capacidad de búsqueda de secuencias de las herramientas). Se consiguen métodos más generales tanto de fuentes comerciales (como “FrameSearch”, distribuido como parte del paquete Accelrys GCG), como de software de código abierto (como Genewise).
El método de programación dinámica garantiza encontrar un alineamiento óptimo dada una función de puntuación en particular; sin embargo, identificar una buena función de puntuación es, usualmente, más una cuestión empírica que teórica. A pesar de que la programación dinámica es extensible a más de dos secuencias, es prohibitivamente lenta para un alto número de secuencias extremadamente largas.
Métodos de palabra corta[editar]
Los métodos de palabra corta, también conocidos como métodos de k-tuplas, son métodos heurísticos que no garantizan encontrar una solución de alineamiento óptima, pero son significativamente más eficientes que la programación dinámica. Estos métodos son especialmente útiles en búsquedas sobre bases de datos a gran escala, donde se asume que una larga proporción de las secuencias candidatas no tendrán coincidencias significativas con la secuencia problema. Los métodos de palabra corta son más conocidos por su implementación en las herramientas de búsqueda en bases de datos FASTA y la familia BLAST.4 Estos métodos identifican en la secuencia problema una serie de subsecuencias cortas que no se solapan (“palabras”), y que se contrastan contra las secuencias de la base de datos. Las posiciones relativas de la palabra en las dos secuencias a comparar se restan para obtener un valor de desplazamiento; se manifestará así una región de alineamiento si varias palabras diferentes producen el mismo desplazamiento. Sólo si esta región es detectada, estos métodos aplicarán criterios de alineamiento más sensibles. De esta forma se eliminan muchas comparaciones innecesarias entre secuencias de similitud inapreciable.
En el método FASTA, el usuario define un valor “k” para definir la longitud de la palabra con la cual buscar en la base de datos. El método es más lento, pero más sensible, para valores bajos de “k”, que también son preferibles para búsquedas que impliquen una secuencia problema muy corta. La familia BLAST de métodos de búsqueda proporciona varios algoritmos optimizados para tipos particulares de problemas, tales como la búsqueda de coincidencias entre secuencias escasamente relacionadas. BLAST fue desarrollado para proporcionar una alternativa más rápida a FASTA sin sacrificar demasiada precisión. Como FASTA, BLAST utiliza una palabra de búsqueda de longitud “k”, pero sólo evalúa las coincidencias más significativas de las palabras, en lugar de cada coincidencia como hace FASTA. La mayoría de las implementaciones de BLAST usan una longitud de palabra fijada por defecto que se optimiza para el problema y el tipo de base de datos, y que se cambia sólo bajo circunstancias específicas tales como búsquedas con secuencias problema repetitivas o muy cortas. Pueden encontrarse implementaciones a través de varios portales web, como EMBL FASTA y NCBI BLAST.
Alineamiento múltiple de secuencias[editar]

{kind=link}
{kind=link}
El alineamiento múltiple de secuencias es una extensión del alineamiento de pares que incorpora más de dos secuencias al mismo tiempo. Los métodos de alineamiento múltiple intentan alinear todas las secuencias de un conjunto dado. Los alineamiento múltiples son usados a menudos en la identificación de regiones conservadas en un grupo de secuencias que hipotéticamente están relacionadas evolutivamente. Estos motivos conservados pueden ser usados en conjunto con la estructura y con información mecanística para localizar sitios activoscatalíticos de las enzimas. Los alineamientos son también utilizados para ayudar al establecimiento de relaciones evolutivas mediante la construcción de árboles filogenéticos. Los alineamientos múltiples de secuencias son computacionalmente difíciles de producir y la mayoría de las formulaciones del problema conducen a problemas de optimización combinatorial NP-completos.5 Sin embargo, la utilidad de estos alineamientos en la bioinformática ha dado lugar al desarrollo de una variedad de métodos adecuados para la alineación de tres o más secuencias.
Programación dinámica[editar]
La técnica de programación dinámica es teóricamente aplicable a cualquier número de secuencias; sin embargo, y puesto que es computacionalmente costosa tanto en tiempo como en memoria, raramente se usa en su forma más básica para más de tres o cuatro secuencias. Este método requiere la construcción de un equivalente n-dimensional a la matriz formada por dos secuencias, donde “n” es el número de secuencias problema. La programación dinámica estándar se usa primero en todos los emparejamientos entre las secuencias problema, por lo que el “espacio de alineamiento” se rellena considerando posibles coincidencias o huecos en las posiciones intermedias, construyendo, finalmente y en esencia, un alineamiento entre cada alineamiento de dos secuencias. Aunque esta técnica es computacionalmente costosa, su garantía de una solución global óptima es útil en casos donde sólo unas cuantas secuencias necesitan ser alineadas con precisión. Un método para reducir las demandas computacionales de programación dinámica, que depende de la función objetivo “suma de pares”, ha sido implementado en el paquete de software MSA.6
Métodos progresivos[editar]
Los método progresivos, jerárquicos, o por árbol, generan un alineamiento múltiple de secuencias alineando primero las secuencias más similares, para ir añadiendo sucesivamente al alineamiento secuencias o grupos menos relacionados, hasta que el conjunto problema completo ha sido incorporado a la solución. El árbol inicial describiendo el parentesco de las secuencias se basa en comparaciones de emparejamientos que podrían incluir métodos de emparejamiento heurístico para alineamientos similares a FASTA. Los resultados del alineamiento progresivo dependen de la elección de las secuencias “más relacionadas”, por lo que pueden ser sensibles a imprecisiones en los alineamientos de emparejamientos iniciales. La mayoría de los métodos progresivos de alineamiento múltiple de secuencias ponderan adicionalmente las secuencias en el conjunto problema de acuerdo a su parentesco, lo que reduce la probabilidad de efectuar una pobre elección de las secuencias iniciales y así se mejora la precisión del alineamiento.
Un buen número de variaciones de la implementación progresiva de Clustal789 se utilizan para alineamientos múltiples de secuencias, construcción de árboles filogenéticos, y como entrada para la predicción de estructura de proteínas. Una variante del método progresivo más lenta pero más precisa se conoce como “T-Coffee” (Tree-based Consistency Objective Function For alignment Evaluation),10 de la que pueden encontrarse implementaciones en ClustalW y T-Coffee.
Métodos iterativos[editar]
Los métodos iterativos intentan mejorar el punto débil de los métodos progresivos: su fuerte dependencia de la precisión de los alineamientos de los emparejamientos iniciales. Los métodos iterativos optimizan una función objetivo basada en un método seleccionado de puntuación de alineamiento mediante la asignación de un alineamiento global inicial y el posterior realineamiento de subconjuntos de secuencias. Los subconjuntos realineados son entonces alineados consigo mismos para producir la siguiente iteración de alineamiento múltiple de secuencias. Se encuentran bajo análisis varias formas de selección de los subgrupos de secuencias y de la función objetivo.11
Descubrimiento de motivos[editar]
Para el descubrimiento de motivos, o análisis de perfiles, se construyen alineamientos múltiples globales de secuencias que intentan alinear secuencias motivo cortas conservadas entre las secuencias del conjunto problema. Se hace, usualmente, construyendo primero un alineamiento múltiple de secuencias global, tras el cual las regiones altamente conservadas se identifican y se utilizan para construir un conjunto de matrices de perfil (también denominadas matrices ponderadas o matrices de pesos). La matriz del perfil de cada región conservada se dispone como una matriz de puntuación, pero sus cifras de frecuencia para cada aminoácido o nucleótido en cada posición se derivan de la distribución de los caracteres de la región conservada, en lugar de una distribución empírica más general. Las matrices de perfil se usan para buscar ocurrencias del motivo que caracterizan en otras secuencias. En los casos en los que el conjunto de datos original contenga un pequeño número de secuencias, o sólo secuencias muy relacionadas, se añaden pseudocontadores para normalizar las distribuciones de caracteres representadas en el motivo.
Técnicas inspiradas por las ciencias de la computación[editar]
Una variedad de algoritmos generales de optimización usados comúnmente en ciencias de la computación se han aplicado también al problema del alineamiento de secuencias. Los modelos ocultos de Márkov se han utilizado para producir registros de probabilidades para una familia de posibles alineamientos múltiples de secuencias sobre un conjunto problema dado. Aunque los primeros métodos basados en estos modelos eran de rendimiento poco brillante, aplicaciones posteriores los han encontrado especialmente efectivos para detectar secuencias remotamente relacionadas, puesto que son menos susceptibles al ruido creado por sustituciones conservativas o semiconservativas.12 Los algoritmos genéticos y el simulated annealing se han usado para optimizar las puntuaciones de alineamientos múltiples de secuencias, valorándolos mediante una función de puntuación como la del método de suma de pares.
Alineamiento estructural[editar]
Los alineamientos estructurales, que son específicos de las proteínas y, algunas veces, de secuencias de ARN, usan información sobre la estructura secundaria y terciaria de la proteína o molécula de ARN como ayuda para alinear las secuencias. Estos métodos pueden usarse para dos o más secuencias, y producen típicamente alineamientos locales. Sin embargo, y puesto que dependen de la disponibilidad de información estructural, sólo pueden ser usados para secuencias cuyas correspondientes estructuras sean conocidas (a través, normalmente, de cristalografía de rayos X o espectroscopia de resonancia magnética nuclear). Puesto que la estructura tanto de proteínas como de ARN está más conservada evolutivamente que su secuencia,13 los alineamientos estructurales pueden ser más fidedignos entre secuencias que estén muy lejanamente relacionadas y que hayan divergido tan extensamente que la comparación de las secuencias no pueda detectar fehacientemente su similitud.
Los alineamientos estructurales se usan como el “patrón oro” para evaluar alineamientos en la predicción de estructura de proteínas basada en homología14 ya que explícitamente alinean regiones de la secuencia de la proteína que son estructuralmente similares en lugar de depender exclusivamente en la información derivada de la secuencia. No obstante, los alineamientos estructurales no pueden usarse en la predicción de la estructura puesto que al menos una secuencia en el conjunto problema es el objetivo a modelar, para el cual la estructura se desconoce. Se ha demostrado que, dado el alineamiento estructural entre un objetivo y una secuencia molde, se pueden producir modelos altamente precisos de la proteína objetivo. Un importante obstáculo en la predicción de la estructura basada en homología es la producción de alineamientos estructuralmente precisos dada sólo información de la secuencia.14
DALI[editar]
El método DALI (del inglés, Distance matrix ALIgnment, alineamiento de matriz de distancias), es un método fragmentario para construir alineamientos estructurales basados en contactar patrones de similitud entre sucesivos hexapéptidos en las secuencias problema.15 Puede generar emparejamientos o alineamientos múltiples, e identificar los vecinos estructurales de una secuencia problema en el Protein Data Bank (PDB). Se ha usado para construir la base de datos de alineamientos estructurales FSSP (del inglés Families of Structurally Similar Proteins, familias de proteínas estructuralmente similares). Puede accederse a un servidor web de DALI en EBI DALI , y la FSSP se localiza en la base de datos DALI.
SSAP[editar]
SSAP (del inglés Sequential Structure Alignment Program, programa de alineamiento de estructura secuencial) es un método de alineamiento estructural basado en programación dinámica que usa vectores “átomo a átomo” en el espacio de la estructura como puntos a comparar. Se ha extendido desde su descripción original para incluir tanto alineamientos múltiples como emparejamientos,16 y se ha usado en la construcción del CATH (del inglés Class, Architecture, Topology, Homology; clase, arquitectura, topología, homología), base de datos jerárquica de clasificación de plegamientos de proteínas.17 La base de datos CATH puede accederse en la Clasificación de Estructura de Proteínas CATH.
Extensión combinatoria[editar]
El método de extensión combinatoria para alineamiento estructural genera un alineamiento estructural de pares usando geometría local para alinear fragmentos cortos de las dos proteínas a analizar, y juntar entonces estos fragmentos en un alineamiento mayor.18 Basado en medidas tales como la raíz del error cuadrático medio en superposición de proteínas como sólidos rígidos, distancias entre residuos, estructura secundaria local, y características medioambientales circundantes tales como la hidrofobicidad de los residuos vecinos, se generan alineamientos locales llamados “pares de fragmentos alineados” que se usan para construir una matriz de similitud representando todos los alineamientos estructurales posibles dentro de un criterio de corte predefinido. Se traza, entonces, una trayectoria desde un estado de la estructura de una proteína a otro a través de la matriz, extendiendo el creciente alineamiento un fragmento cada vez. La trayectoria óptima define el alineamiento por extensión combinatoria. Un servidor web que implementa el método y proporciona una base de datos de emparejamientos de estructura en el Protein Data Bank se localiza en el sitio de Combinatorial Extension.
Análisis filogenético[editar]
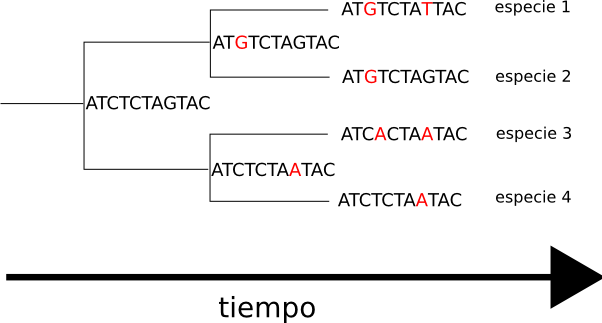
La filogenia y el alineamiento de secuencias son campos íntimamente relacionados debido a su necesidad compartida de evaluar el parentesco entre secuencias. La filogenia hace un uso extensivo de los alineamientos de secuencias en la construcción e interpretación de árboles filogenéticos, que se usan para clasificar las relaciones evolutivas entre genes homólogos representados en el genoma de especies divergentes. El grado en el que las secuencias de un conjunto problema difieren está relacionado cualitativamente con la distancia evolutiva entre ellas. De forma simplificada, una alta identidad de secuencias sugiere que tienen un comparativamente joven ancestro común más reciente, mientras que una baja identidad sugiere que la divergencia es más remota. Esta aproximación, que refleja la hipótesis de “reloj molecular” (hipótesis que asume un ritmo aproximadamente constante de cambio evolutivo, que puede utilizarse para extrapolar el tiempo transcurrido desde la primera divergencia de dos genes -o tiempo de “coalescencia”-), asume que los efectos de la mutación y de la selección natural son constantes a lo largo de linajes de secuencias. No toma en cuenta, por lo tanto, posibles diferencias entre organismos o especies en los ritmos de reparación del ADN, o la posible conservación funcional de regiones específicas en una secuencia. (En el caso de secuencias de nucleótidos, la hipótesis de reloj molecular en su forma más básica también deja de lado la diferencia en las tasas de aceptación entre mutaciones silenciosas, que no alteran el significado de un determinado codón, y otras mutaciones que resultan en la incorporación de un aminoácido diferente en la proteína.) Métodos con mayor precisión estadística permiten variar el ritmo evolutivo en cada rama del árbol filogenético, produciendo así mejores estimaciones de los tiempos de coalescencia de los genes.
Las técnicas de alineamiento múltiple progresivo producen un árbol filogenético necesariamente, ya que van incorporando secuencias en el creciente alineamiento según su orden de parentesco. Otras técnicas que reúnen alineamientos múltiples de secuencias y árboles filogenéticos, puntúan y ordenan los árboles en primer lugar, y calculan después un alineamiento múltiple de secuencias a partir del árbol de mayor puntuación. Los métodos comunes de construcción de árboles filogenéticos son principalmente heurísticos puesto que el problema de seleccionar el árbol óptimo, al igual que el problema de seleccionar el alineamiento múltiple de secuencias óptimo, es NP-complejo.19
Valoración de su significación[editar]
Los alineamientos de secuencias son útiles en bioinformática para identificar similitudes entre secuencias, producir árboles filogenéticos, y desarrollar modelos de homología sobre estructuras de proteínas. Sin embargo, la relevancia biológica de los alineamientos no siempre es clara. Se asume a menudo que los alineamientos reflejan un grado de cambio evolutivo entre secuencias que descienden de un ancestro común; pero es formalmente posible que la convergencia evolutiva pueda darse para producir similitudes aparentes entre proteínas que no estén evolutivamente relacionadas, pero que lleven a cabo funciones similares y tengan parecidas estructuras.
En búsquedas en bases de datos, como con BLAST, los métodos estadísticos pueden determinar la probabilidad de un alineamiento particular casual entre secuencias, o regiones de secuencias, dado el tamaño y la composición de la base de datos en cuestión. Estos valores pueden variar significativamente dependiendo del espacio de búsqueda. En particular, la probabilidad de encontrar por casualidad un alineamiento dado se incrementa si la base de datos consiste sólo en secuencias del mismo organismo que la secuencia problema. Secuencias repetitivas en la base de datos o en la consulta también pueden distorsionar tanto la búsqueda de resultados y la valoración de su significación estadística. BLAST filtra automáticamente tales secuencias repetitivas en la consulta para evitar éxitos aparentes que correspondan a artefactos estadísticos.
Funciones de puntuación[editar]
Para producir buenos alineamientos es importante la elección de una función de puntuación que refleje observaciones biológicas o estadísticas sobre secuencias conocidas. Las secuencias de proteínas son alineadas usando frecuentemente matrices de sustitución que reflejan las probabilidades de particulares sustituciones carácter por carácter. Una serie de matrices denominadas matrices PAM (del inglés Point Accepted Mutation, mutación puntual aceptada, originalmente definidas por Margaret Dayhoff, por lo que a veces se denominan matrices Dayhoff) codifican explícitamente las aproximaciones evolutivas considerando las frecuencias y probabilidades de mutaciones particulares de aminoácidos. Otra serie común de matrices de puntuación, conocidas como BLOSUM (del inglés Blocks Substitution Matrix, matriz de sustitución de bloques), codifica probabilidades de sustitución derivadas empíricamente. Se utilizan variantes de ambos tipos de matrices para detectar secuencias con diferentes niveles de divergencia, permitiendo así a los usuarios de BLAST o FASTA restringir sus búsquedas a coincidencias más cercanamente relacionadas, o extenderlas para detectar secuencias más divergentes. Las penalizaciones por gaps representan la introducción de huecos (en el modelo evolutivo, una mutación por inserción o borrado) en secuencias tanto de nucleótidos como de proteínas, y estos valores de penalización, por lo tanto, deberían ser proporcionales a la frecuencia esperada de tales mutaciones. La calidad de los alineamientos producidos depende, en consecuencia, de la calidad de la función de puntuación.
Puede resultar muy útil e instructivo intentar el mismo alineamiento diferentes veces con diferentes elecciones de matrices de puntuación y/o diferentes valores de penalización por huecos, y comparar los resultados. Las regiones donde la solución sea poco consistente, o no sea única, pueden ser identificadas a menudo observando qué regiones del alineamiento son robustas a variaciones en los parámetros de alineación.
Usos no biológicos[editar]
Los métodos usados para alineamientos de secuencias biológicas pueden también encontrar aplicaciones en otros campos, y muy notablemente en el procesamiento de lenguajes naturales. Las técnicas que generan el conjunto de elementos desde el que las palabras se seleccionarán en los algoritmos de generación de lenguajes naturales han pedido prestadas técnicas de alineamiento de secuencias a la bioinformática para producir versiones lingüísticas de pruebas matemáticas generadas por ordenador.20 En el campo de lingüística histórica y comparativa, se ha usado el alineamiento de secuencias para automatizar parcialmente el método comparativopor el que tradicionalmente los lingüistas reconstruyen lenguajes.21 También se han aplicado técnicas de alineamiento de secuencias en la investigación de negocios y marketing analizando series temporales de compras.22
Software[editar]
Las herramientas software comunes usadas para tareas generales de alineamiento de secuencias incluyen ClustalW y T-coffee para el alineamiento, y BLAST para búsquedas en bases de datos. Una lista mucho más completa de software disponible, categorizado por algoritmo y tipo de alineamiento, puede encontrarse en el Anexo:Software para alineamiento de secuencias.
Los algoritmos de alineamiento y el software pueden ser contrastados directamente usando un conjunto estandarizado de benchmark de referencia para alineamientos múltiples de secuencias denominado BAliBASE.23 El conjunto de datos consiste en alineamientos estructurales que pueden ser considerados como un estándar contra el cual se comparan los métodos basados en secuencias. Ha sido tabulado el rendimiento relativo de bastantes métodos comunes de alineamiento encontrados frecuentemente en problemas de alineación, y los resultados más significativos se han publicado en línea en BAliBASE.24 En el banco de trabajo de proteínas STRAP puede computarse una detallada lista de puntuaciones de BAliBASE para varias herramientas diferentes de alineamiento.
No hay comentarios:
Publicar un comentario