La deformación se refiere a un conjunto de procedimientos analíticos utilizados para separar e identificar componentes individuales de una sustancia química formulada. [1] [2] [3] [4] La deformación aplica métodos de química analítica y se usa a menudo para obtener inteligencia competitiva sobre productos químicos. La deformación está relacionada con la ingeniería inversa ; sin embargo, este último concepto está más estrechamente asociado con los procedimientos utilizados para descubrir los principios de funcionamiento de un dispositivo o un sistema diseñado mediante el examen y desmontaje de su estructura. El término, ingeniería inversa, se ha vinculado específicamente y casi exclusivamente al campo de la ingeniería de software; [5] [6]mientras que, la deformulación es un término más aplicable al campo de la fabricación química. La deformación de una mezcla química multicomponente puede ocurrir en varios contextos, incluida la investigación de las causas de la falla de un producto químico, la evaluación comparativa competitiva, la investigación legal para obtener evidencia de infracción de patente o la investigación y desarrollo de nuevos productos. Dependiendo de este contexto y del nivel de información que se busque, los requisitos de los análisis de deformulación pueden diferir. [7] Los procesos de deformación generalmente requieren la aplicación de varios métodos analíticos, y la selección de los métodos depende del grado de confianza requerido en los resultados. Los métodos de deformulación también tienen similitud con los métodos de la química forense. en el cual se pueden aplicar procedimientos analíticos para descubrir las causas de fallas materiales o para resolver una cuestión legal.
Deformulación relacionada con los derechos de propiedad intelectual [ editar ]
En los Estados Unidos, la ley federal reconoce una práctica legal para el estudio de un artículo con la esperanza de obtener una comprensión detallada de la forma en que funciona con el fin de crear productos duplicados o superiores sin el beneficio de tener los planes para el original. ít. El artículo estudiado primero debe haber sido obtenido legalmente, no robado o malversado de otra manera. [8]El propósito de la protección de la propiedad intelectual es proporcionar incentivos para invertir y promover el conocimiento colectivo. Se considera que la deformación o la ingeniería inversa ayudan a educar y promover una competencia sana. Se considera una herramienta de aprendizaje que proporciona un camino para crear productos nuevos y competitivos con un mejor desempeño y menor costo que lo que existe actualmente en el mercado. La deformación a menudo se considera junto con la evaluación comparativa, el mapeo de patentes y otros procesos de recopilación de inteligencia de la competencia como un medio para llevar a cabo las actividades diarias. [9]
Otros países pueden tener diferentes concepciones sobre los derechos de propiedad intelectual y sobre las concesiones legales para la deformación o la ingeniería inversa de los artículos. Para obtener información sobre el estado legal de las prácticas de deformación en otros países del mundo, es recomendable consultar con un experto en derecho de propiedad intelectual.
Procedimientos deformulación [ editar ]
Se puede realizar un análisis preliminar de orden cero para responder preguntas fundamentales sobre la naturaleza del material desconocido. Los métodos que podrían usarse para el análisis preliminar incluyen métodos espectroscópicos, como la espectroscopia infrarroja o la espectroscopia de fluorescencia de rayos X. Los resultados de la caracterización del orden cero del material informan las elecciones posteriores en las etapas posteriores del análisis.
Una mezcla química formulada puede contener varias fases, como material suspendido o emulsionado. Un análisis de primer orden del material puede implicar la separación de fases. La centrifugación , extracción y filtración son ejemplos de métodos que separan material en diferentes fases. La centrifugación es efectiva para separar fases que difieren en densidad. La extracción es efectiva para separar fases líquidas inmiscibles. La filtración es efectiva para separar partículas dispersas que son lo suficientemente grandes en tamaño para ser atrapadas en un filtro. Esta separación inicial puede requerir la selección de disolventes apropiados para disolver los componentes sólidos o actuar como un diluyente para líquidos. La determinación cuantitativa de las fases a menudo se determina gravimétricamente.
Una vez separadas, cada fase del material es en sí misma una mezcla química para ser analizada más a fondo. Un análisis de segundo orden de cada fase generalmente implicará una selección entre los métodos analíticos disponibles para separar aún más estos componentes. Los métodos analíticos utilizados en las fases líquidas pueden incluir la destilación o uno de una variedad de métodos de separación cromatográfica. La destilaciónsepara los componentes de una mezcla líquida de acuerdo con las diferencias en sus puntos de ebullición. La cromatografía separa los componentes de una mezcla líquida o gaseosa de acuerdo con las diferencias en el tiempo de retención a medida que la mezcla interactúa con una fase estacionaria. Los componentes individuales así separadas se pueden identificar entonces por una variedad de métodos de detección, incluyendo la espectroscopia de infrarrojos , espectroscopia Raman, espectrometría de masas y espectrometría de resonancia magnética nuclear . Los métodos utilizados para analizar más a fondo los sólidos pueden incluir el análisis térmico (como el análisis termogravimétrico o la calorimetría diferencial de barrido ), la difracción de rayos X para caracterizar los sólidos cristalinos, microscopía, pirólisis , análisis de combustión o métodos espectroscópicos de superficie.
En algunos contextos, pueden requerirse etapas adicionales de análisis de los componentes separados. Los ingredientes activos de un producto químico formulado que lo diferencian de otro material similar pueden incluir ingredientes patentados o aditivos funcionales específicos. [10] Tales ingredientes que juegan un papel clave en el rendimiento del material en una aplicación pueden requerir un análisis de tercer orden para caracterizarlos más completamente. Algunos ejemplos de aditivos funcionales incluyen surfactantes , emulsionantes , dispersantes , promotores de adhesión, agentes de nivelación, colorantes y pigmentos , antioxidantes , conservantes., y abrillantadores ópticos. Prácticamente todos los tipos de productos formulados químicamente están asociados con su propio formulario de posibles opciones de aditivos funcionales que pueden desempeñar un papel crítico en el desempeño. Por lo tanto, la deformación puede requerir tanto un desglose de la composición del material como la identificación del papel funcional de los ingredientes clave.
Ejemplos de tipos de productos químicos y tipos de aditivos funcionales [ editar ]
| Producto químico formulado | Aditivos funcionales posibles | Referencias |
|---|---|---|
| Detergente de lavandería | Tensioactivos, agentes blanqueadores, desespumantes, enzimas, inhibidores de la corrosión, fragancias, agentes espesantes. | [11] |
| Tinta litográfica offset | Secadores, ceras, antioxidantes, modificadores reológicos, aditivos para litografía. | [12] [13] |
| Pintura de la casa interior | Pigmentos, extensores, iniciadores, agentes de transferencia de cadena, agentes coalescentes, agentes humectantes, estabilizadores de congelación y descongelación. | [14] [15] |
| Adhesivo de laminación | Estabilizador coloidal, surfactantes aniónicos, surfactantes no iónicos, agentes de transferencia de cadena, plastificantes, humectantes. | [dieciséis] |
| Aceite de motor automotriz | Depresores del punto de fluidez, modificadores de la viscosidad, antioxidantes, inhibidores de detergentes, aditivos antidesgaste, modificadores de fricción | [17] |
| Máscara para soldar | Fotoiniciadores, diluyentes reactivos. | [18] |
| Bebida carbonatada | Conservantes, acidulantes, edulcorantes. | [19] |
La determinación analítica de un aditivo funcional tiene problemas particulares asociados. La concentración de un aditivo funcional puede ser baja en comparación con otros ingredientes; por lo tanto, puede ser difícil de detectar. Los ingredientes patentados son especialmente difíciles de identificar correctamente. El papel funcional de un componente clave puede no ser obvio en la inspección. Un ingrediente clave puede no ser revelado por el fabricante del material, pero debe mantenerse como un secreto comercial . El estudio cuidadoso de la literatura comercial y las solicitudes de patentes asociadas con el fabricante puede ayudar al analista en la caracterización.
límite de detección , límite inferior de detección , o LOD (límite de detección), es la cantidad más baja de una sustancia que puede ser distinguida de la ausencia de dicha sustancia (un valor en blanco ) con un declarado nivel de confianza (generalmente 99%). [1] [2] El límite de detección se estima a partir de la media del blanco, la desviación estándar del blanco y algún factor de confianza. Otra consideración que afecta el límite de detección es la precisión del modelo utilizado para predecir la concentración a partir de la señal analítica sin procesar.
Hay una serie de diferentes "límites de detección" que se utilizan comúnmente. Estos incluyen el límite de detección del instrumento ( IDL ), el límite de detección del método ( MDL ), el límite de cuantificación práctica ( PQL ) y el límite de cuantificación ( LOQ ). Incluso cuando se usa la misma terminología, puede haber diferencias en el LOD según los matices de qué definición se usa y qué tipo de ruido contribuye a la medición y calibración. [3]
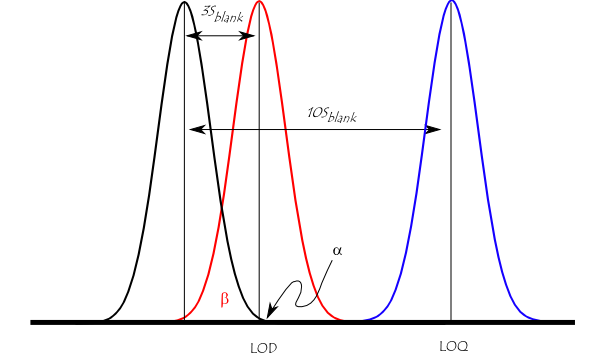
La siguiente figura ilustra la relación entre el blanco, el límite de detección (LOD) y el límite de cuantificación(LOQ) al mostrar la función de densidad de probabilidad para mediciones normalmente distribuidas en el blanco, en el LOD definido como 3 * desviación estándar de el blanco, y en el LOQ definido como 10 * desviación estándar del blanco. Para una señal en el LOD, el error alfa (probabilidad de falsos positivos) es pequeño (1%). Sin embargo, el error beta (probabilidad de un falso negativo) es del 50% para una muestra que tiene una concentración en el LOD (línea roja). Esto significa que una muestra podría contener una impureza en el LOD, pero hay un 50% de probabilidad de que una medición dé un resultado menor que el LOD. En el LOQ (línea azul), hay una probabilidad mínima de un falso negativo.
Límite de detección de instrumentos [ editar ]
La mayoría de los instrumentos analíticos producen una señal incluso cuando se analiza un blanco ( matriz sin analito). Esta señal se conoce como el nivel de ruido. La IDL es la concentración de analito que se requiere para producir una señal mayor a tres veces la desviación estándar del nivel de ruido. Esto puede medirse en la práctica analizando 8 o más estándares en el IDL estimado y luego calculando la desviación estándar de las concentraciones medidas de esos estándares. El límite de detección (según IUPAC) es la concentración más pequeña o la cantidad absoluta de analito que tiene una señal significativamente mayor que la señal que surge de un blanco de reactivo. Matemáticamente, la señal del analito en el límite de detección (Sdl) viene dada por: .

donde Sreag es la señal para un blanco de reactivo, σreag es la desviación estándar conocida para la señal del blanco de reactivo.
También se han desarrollado otros enfoques para definir el límite de detección. En la espectrometría de absorción atómica, por lo general, el límite de detección se determina para un elemento determinado mediante el análisis de una solución diluida de este elemento y el registro de las absorbancias correspondientes. El experimento se repite 10 veces. El 3σ de la señal de absorbancia registrada puede considerarse como el límite de detección para el elemento específico en las condiciones experimentales utilizadas: longitud de onda, tipo de llama, instrumento.
Método de detección de límite [ editar ]
A menudo hay más en el método analítico que solo realizar una reacción o enviarlo a un análisis directo. Por ejemplo, podría ser necesario calentar una muestra que se analizará para un metal en particular con la adición de ácido primero (esto se llama digestión ). La muestra también se puede diluir o concentrar antes del análisis en un instrumento. Los pasos adicionales en un análisis agregan oportunidades adicionales para el error. Dado que los límites de detección se definen en términos de error, esto naturalmente aumentará el límite de detección medido. Este límite de detección (con todos los pasos del análisis incluidos) se denomina MDL. El método práctico para determinar el MDL es analizar 7 muestras de concentración cerca del límite esperado de detección. La desviación estándarEntonces se determina. La distribución t de un solo lado se determina y se multiplica frente a la desviación estándar determinada . Para siete muestras (con seis grados de libertad), el valor de t para un nivel de confianza del 99% es 3.14. En lugar de realizar el análisis completo de siete muestras idénticas, si se conoce el límite de detección del instrumento, la MDL se puede estimar multiplicando el límite de detección del instrumento o el nivel inferior de detección por la dilución antes de analizar la solución de muestra en el instrumento. Esta estimación, sin embargo, ignora cualquier incertidumbre que surja al realizar la preparación de la muestra y, por lo tanto, probablemente subestime la verdadera MDL.
El límite de cuantificación [ editar ]
El LOQ es el límite en el cual la diferencia entre dos valores distintos se puede discernir razonablemente. El LOQ puede ser drásticamente diferente entre los laboratorios, por lo que comúnmente se usa otro límite de detección que se conoce como el Límite de Cuantificación Práctica (PQL).
Las constantes de equilibrio se determinan para cuantificar los equilibrios químicos . Cuando una constante de equilibrio K se expresa como un cociente de concentración,
![{\ displaystyle K = {\ frac {\ mathrm {[S]} ^ {\ sigma} \ mathrm {[T]} ^ {\ tau} \ cdots} {\ mathrm {[A]} ^ {\ alpha} \ mathrm {[B]} ^ {\ beta} \ cdots}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7fbf3623d6284c219011f318e2197779ab194931)
se implica que el cociente de actividad es constante. Para que esta suposición sea válida, las constantes de equilibrio deben determinarse en un medio de fuerza iónica relativamente alta . Cuando esto no sea posible, se debe considerar la posible variación de la actividad.
La expresión de equilibrio anterior es una función de las concentraciones [A], [B], etc. de la especie química en equilibrio. El valor constante de equilibrio puede determinarse si se puede medir cualquiera de estas concentraciones. El procedimiento general es que la concentración en cuestión se mide para una serie de soluciones con concentraciones analíticas conocidas de los reactivos. Típicamente, una titulación se realiza con uno o más reactivos en el recipiente de titulación y uno o más reactivos en la bureta. Al conocer las concentraciones analíticas de los reactivos inicialmente en el recipiente de reacción y en la bureta, todas las concentraciones analíticas se pueden derivar en función del volumen (o masa) del valorante agregado.
Las constantes de equilibrio pueden derivarse ajustando mejor los datos experimentales con un modelo químico del sistema de equilibrio.
Métodos experimentales [ editar ]
Hay cuatro métodos experimentales principales. Para métodos menos utilizados, ver Rossotti y Rossotti. [1]
Mediciones potenciométricas [ editar ]
Una concentración libre [A] o actividad {A} de una especie A se mide por medio de un electrodo selectivo de iones como el electrodo de vidrio . Si el electrodo se calibra utilizando estándares de actividad, se supone que la ecuación de Nernst se aplica en la forma

donde E 0 es el potencial de electrodo estándar . Cuando se utilizan soluciones tampón de pH conocido para la calibración, la lectura del medidor será un pH.

A 298 K, 1 unidad de pH es aproximadamente igual a 59 mV. [2]
Cuando el electrodo se calibra con soluciones de concentración conocida, por medio de una fuerte titulación ácido-fuerte, por ejemplo, se supone una ecuación de Nernst modificada.
![{\ displaystyle E = E ^ {0} + s \ log _ {10} \ mathrm {[A]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c404b52bc56775c11adfd79fab6c73166abc0193)
donde s es un factor de pendiente empírico. Se puede preparar una solución de concentración conocida de iones de hidrógeno mediante la estandarización de un ácido fuerte contra el bórax . El ácido clorhídrico de ebullición constante también se puede usar como un estándar primario para la concentración de iones de hidrógeno.
Medidas espectrofotométricas [ editar ]
Absorbancia [ editar ]

donde l es la longitud del camino óptico, ε es una absorbancia molar a la longitud del camino unitario y c es una concentración. Más de una de las especies puede contribuir a la absorbancia. En principio, la absorbancia se puede medir solo en una longitud de onda, pero en la práctica actual es común registrar espectros completos.
Fluorescencia (luminiscencia) intensidad [ editar ]
Se supone que la intensidad de la luz dispersada es una función lineal de las concentraciones de las especies.

donde φ es una constante de proporcionalidad.
Mediciones de desplazamiento químico de RMN [ editar ]
Se supone que el intercambio químico es rápido en la escala de tiempo de RMN. Un cambio químico individual δes el promedio ponderado en fracciones molares de los cambios δ de núcleos en las especies contribuyentes.

Ejemplo: la p K a del grupo hidroxilo en ácido cítrico se ha determinado a partir de los datos de cambio químico de 13 C como 14.4. Para esta determinación no se pudo utilizar potenciometría ni espectrofotometría UV visible . [3]
Mediciones calorimétricas [ editar ]
La medición simultánea de K y Δ H para aductos 1: 1 se realiza de forma rutinaria mediante calorimetría de titulación isotérmica . La extensión a sistemas más complejos está limitada por la disponibilidad de software adecuado.
Rango y limitaciones [ editar ]
- Potenciometría
- El electrodo más utilizado es el electrodo de vidrio, que es selectivo para el ion hidrógeno. Esto es adecuado para todos los equilibrios ácido-base . Los valores log 10 β entre aproximadamente 2 y 11 se pueden medir directamente mediante valoración potenciométrica utilizando un electrodo de vidrio . Este enorme rango es posible debido a la respuesta logarítmica del electrodo. Las limitaciones surgen porque la ecuación de Nernst se descompone a un pH muy bajo o muy alto. El rango se puede ampliar utilizando el método de competición. Un ejemplo de la aplicación de este método se puede encontrar en el cianuro de paladio (II) .
Cuando se usa un electrodo de vidrio para obtener las mediciones de las que dependen las constantes de equilibrio calculadas, la precisión de los parámetros calculados está limitada por efectos secundarios tales como la variación de los potenciales de unión líquida en el electrodo. En la práctica, es prácticamente imposible obtener una precisión para log β mejor que ± 0.001. - Absorbancia y luminiscencia
- Generalmente se cita un límite superior en el log 10 β de 4, que corresponde a la precisión de las mediciones, pero también depende de la intensidad del efecto. Los espectros de las especies contribuyentes deben ser claramente distintos entre sí.
- RMN
- La precisión limitada de las mediciones de desplazamiento químico también pone un límite superior de aproximadamente 4 en log 10 β . Limitado a los sistemas diamagnéticos. La 1 H NMR no se puede usar con soluciones de compuestos en 1 H 2 O.
- Calorimetría
- Actualmente no se dispone de pruebas suficientes.
Métodos computacionales [ editar ]
Se supone que los datos experimentales recopilados comprenden un conjunto de puntos de datos. En cada i-ésimo punto de datos, las concentraciones analíticas de los reactivos, T A ( i ) , T B ( i ) etc. son conocidos junto con una cantidad medida, y i , que depende de uno o más de estas concentraciones analíticas. Un procedimiento computacional general tiene cuatro componentes principales:
- Definición de un modelo químico de los equilibrios.
- Cálculo de las concentraciones de todas las especies químicas en cada solución.
- Refinamiento de las constantes de equilibrio.
- Selección de modelo
El modelo químico [ editar ]
El modelo químico consiste en un conjunto de especies químicas presentes en solución, tanto los reactivosagregados a la mezcla de reacción como las especies complejas formadas a partir de ellos. Denotando los reactivos por A, B ..., cada especie compleja está especificada por los coeficientes estequiométricos que relacionan la combinación particular de reactivos que los forman.
- :

![{\ displaystyle \ beta _ {pq \ cdots} = {\ frac {[{\ ce {A}} _ {p} {\ ce {B}} _ {q} \ cdots]} {[{\ ce {A }}] ^ {p} [{\ ce {B}}] ^ {q} \ cdots}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/41ddfaa95c2aab1288ee0ef5881d7829e498e933)
Cuando se usan programas de computadora de propósito general, es usual usar constantes de asociación acumulativas , como se muestra arriba. Las cargas eléctricas no se muestran en expresiones generales como esta y, a menudo, se omiten en expresiones específicas, por simplicidad de notación. De hecho, las cargas eléctricas no tienen relación con los procesos de equilibrio, salvo que existe un requisito de neutralidad eléctrica general en todos los sistemas.
Con las soluciones acuosas, las concentraciones de protón (ion hidronio) e ion hidróxido están limitadas por la autodisociación del agua.
- :

![{\ displaystyle K _ {\ mathrm {W}} ^ {'} = {\ frac {[H ^ {+}] [OH ^ {-}]} {[H_ {2} O]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/31183901f208f67c181a1cbafa2c7d61b45f73e4)
Con las soluciones diluidas, la concentración de agua se supone constante, por lo que la expresión de equilibrio se escribe en forma del producto iónico del agua.
![{\ displaystyle K _ {\ mathrm {W}} = {\ ce {[H +]}} [{\ ce {OH -}}] \,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/29aa88810dddbd1cfe6122a3c8a3f27fe64d61f4)
Cuando tanto H + como OH - deben considerarse reactivos, uno de ellos se elimina del modelo especificando que su concentración se deriva de la concentración del otro. Normalmente la concentración del ion hidróxido viene dada por
![{\ displaystyle [{\ ce {OH -}}] = {\ frac {K _ {{\ ce {W}}}} {[{\ ce {H +}}]}} \,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c6d83aeaec2578430ca83870deb9a3e00c622e4c)
En este caso, la constante de equilibrio para la formación de hidróxido tiene los coeficientes estequiométricos -1 en relación con el protón y cero para los otros reactivos. Esto tiene implicaciones importantes para todos los equilibrios de protonación en solución acuosa y en particular para las constantes de hidrólisis .
Es bastante habitual omitir en el modelo aquellas especies cuyas concentraciones se consideran insignificantes. Por ejemplo, generalmente se asume que no hay interacción entre los reactivos y / o complejos y el electrolito usado para mantener la fuerza iónica constante o el tampón usado para mantener el pH constante. Estas suposiciones pueden o no estar justificadas. Además, se asume implícitamente que no hay otras especies complejas presentes. Cuando los complejos se ignoran erróneamente, se introduce un error sistemático en los cálculos.
Los valores constantes de equilibrio generalmente se estiman inicialmente por referencia a las fuentes de datos .
Cálculos de especiación [ editar ]
Un cálculo de especiación es uno en el que se calculan las concentraciones de todas las especies en un sistema de equilibrio, conociendo las concentraciones analíticas, T A , T B, etc. de los reactivos A, B, etc. Esto significa resolver un conjunto de ecuaciones no lineales de masa equilibrar
![{\ displaystyle {\ begin {alineado} {\ ce {T_ {A}}} & = [{\ ce {A}}] + \ sum _ {1, nk} p \ beta _ {pq \ cdots} [{ \ ce {A}}] ^ {p} [{\ ce {B}}] ^ {q} \ cdots \\ {\ ce {T_ {B}}} & = [{\ ce {B}}] + \ sum _ {1, nk} q \ beta _ {pq \ cdots} [{\ ce {A}}] ^ {p} [{\ ce {B}}] ^ {q} \ cdots \\ etc. \ fin {alineado}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2006c9c342b8217af7f3ad79e0218ca647bcf264)
para las concentraciones libres [A], [B], etc. Cuando se mide el pH (o emf, E) equivalente, la concentración libre de iones de hidrógeno, [H], se obtiene del valor medido como
![{\ displaystyle [\ mathrm {H}] = 10 ^ {- \ mathrm {pH}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8b22a1dc887dbdb859b455b86ddeea83cdd88106)
![{\ displaystyle [\ mathrm {H}] = 10 ^ {\ mathrm {\ frac {EE ^ {0}} {2.303RT / nF}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/25c8fbe5d77db753c52da389b08f30179d2de934)
y solo se calculan las concentraciones libres de los otros reactivos. Las concentraciones de los complejos se derivan de las concentraciones libres a través del modelo químico.
Algunos autores [4] [5] incluyen los términos reactivos libres en las sumas al declarar constantes β de identidad(unidad) para las cuales los coeficientes estequiométricos son 1 para el reactivo en cuestión y cero para todos los demás reactivos. Por ejemplo, con 2 reactivos, las ecuaciones de balance de masa toman la forma más simple.
![{\ displaystyle {\ begin {alineado} T _ {\ ce {A}} & = \ sum _ {0, nk} p \ beta _ {pq} [{\ ce {A}}] ^ {p} [{\ ce {B}}] ^ {q} \\ [4pt] T _ {\ ce {B}} & = \ sum _ {0, nk} q \ beta _ {pq} [{\ ce {A}}] ^ {p} [{\ ce {B}}] ^ {q} \\\ end {alineado}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c8d8b63491bb40690a8354ffecdfc2ec2e804777)

De esta manera, todas las especies químicas, incluidos los reactantes libres , se tratan de la misma manera, ya que se han formado a partir de la combinación de reactivos que se especifica mediante los coeficientes estequiométricos.
En un sistema de titulación, las concentraciones analíticas de los reactivos en cada punto de titulación se obtienen de las condiciones iniciales, las concentraciones de bureta y los volúmenes. La concentración analítica (total) de un reactivo R en el punto i de titulación viene dada por
![{\ displaystyle T _ {{\ ce {R}}} = {\ frac {{\ ce {R}} _ {0} + v_ {i} {\ ce {[R]}}} {v_ {0} + v_ {i}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5014e528924b4e408f938a3b82cbfb58d71510c0)
donde R 0 es la cantidad inicial de R en el recipiente de titulación, v 0 es el volumen inicial, [R] es la concentración de R en la bureta y v i es el volumen agregado. La concentración de la bureta de un reactivo no presente en la bureta se toma como cero.
En general, la resolución de estas ecuaciones no lineales presenta un desafío formidable debido al enorme rango en el que pueden variar las concentraciones libres. Al principio, los valores para las concentraciones libres deben ser estimados. Luego, estos valores se refinan, generalmente mediante iteraciones de Newton-Raphson . Los logaritmos de las concentraciones libres pueden ser refinados en lugar de las concentraciones libres en sí mismas. El refinamiento de los logaritmos de las concentraciones libres tiene la ventaja adicional de imponer automáticamente una restricción de no negatividad en las concentraciones libres. Una vez que se han calculado las concentraciones de reactante libre, las concentraciones de los complejos se derivan de ellas y las constantes de equilibrio.
Tenga en cuenta que las concentraciones de reactantes libres pueden considerarse como parámetros implícitos en el proceso de refinamiento de la constante de equilibrio. En ese contexto, los valores de las concentraciones libres se ven limitados al obligar a que las condiciones de balance de masa se apliquen en todas las etapas del proceso.
El equilibrio constante refinamiento [ editar ]
El objetivo del proceso de refinamiento es encontrar valores constantes de equilibrio que den el mejor ajuste a los datos experimentales. Esto generalmente se logra minimizando una función objetivo , U , por el método de mínimos cuadrados no lineales . Primero se definen los residuos como

Entonces la función objetivo más general es dada por

La matriz de ponderaciones, W , debería ser, idealmente, la inversa de la matriz de varianza-covarianza de las observaciones. Es raro que esto se sepa. Sin embargo, cuando lo es, el valor esperado de U es uno, lo que significa que los datos se ajustan dentro del error experimental . La mayoría de las veces, solo se conocen los elementos diagonales, en cuyo caso la función objetivo se simplifica a

con W ij = 0 cuando j ≠ i . Los pesos unitarios, W ii = 1 , se usan a menudo, pero, en ese caso, el valor de expectativa de U es el cuadrado medio de la raíz de los errores experimentales.
La minimización se puede realizar utilizando el método de Gauss-Newton . En primer lugar, la función objetivo se linealiza aproximándola como una expansión de la serie de Taylor de primer orden sobre un conjunto de parámetros iniciales, p .

Los incrementos δ p i se agregan a los parámetros iniciales correspondientes, de modo que U es menor que U 0. Como mínimo, los derivados ∂ U∂ p i , que están simplemente relacionados con los elementos de la matriz jacobiana , J

donde p k es el parámetro k th del refinamiento, son iguales a cero. Una o más constantes de equilibrio pueden ser parámetros del refinamiento. Sin embargo, las cantidades medidas (ver arriba) representadas por y no se expresan en términos de las constantes de equilibrio, sino en términos de las concentraciones de las especies, que son funciones implícitas de estos parámetros. Por lo tanto, los elementos jacobianos deben obtenerse mediante la diferenciación implícita .
Los incrementos de los parámetros δ p se calculan resolviendo las ecuaciones normales , derivadas de las condiciones que ∂ U∂ p = 0 como mínimo.

Los incrementos δ p se agregan iterativamente a los parámetros.

donde n es un número de iteración. Las concentraciones de las especies y los valores de calc y se recalculan en cada punto de datos. Las iteraciones se continúan hasta que no se logre una reducción significativa en U , es decir, hasta que se satisfaga un criterio de convergencia. Sin embargo, si los parámetros actualizados no producen una disminución de la función objetivo, es decir, si se produce una divergencia, el cálculo del incremento debe modificarse. La modificación más simple es usar una fracción, f , de incremento calculado, denominado corte de desplazamiento.

En este caso, la dirección del vector de desplazamiento, δ p , no se modifica. Con el algoritmo de Levenberg-Marquardt más poderoso , por otra parte, el vector de desplazamiento se gira hacia la dirección de mayor pendiente , modificando las ecuaciones normales,

donde λ es el parámetro de Marquardt e I es una matriz de identidad. Se han propuesto otros métodos para manejar la divergencia. [5]
Un problema particular surge con la RMN y los datos espectrofotométricos. Para este último, la cantidad observada es absorbancia, A , y la ley de Beer-Lambert se puede escribir como

Se puede ver que, asumiendo que las concentraciones, c, son conocidas, que la absorbancia, A , a una longitud de onda dada,y longitud del camino , es una función lineal de las absorbptividades molares, ε . Con 1 cm de longitud de recorrido, en notación matricial.



Existen dos enfoques para el cálculo de las absortividades molares desconocidas.
- (1) Los valores ε se consideran parámetros de la minimización y el jacobiano se construye sobre esa base. Sin embargo, los valores de ε en sí mismos se calculan en cada paso del refinamiento por mínimos cuadrados lineales:
- usando los valores refinados de las constantes de equilibrio para obtener la especiación. La matriz
- Es un ejemplo de un pseudoinverso .
- Golub y Pereyra [6] mostraron cómo se puede diferenciar la seudoinversión para que los incrementos de los parámetros tanto para la absorción de los molares como para las constantes de equilibrio se puedan calcular resolviendo las ecuaciones normales.
- (2) La ley de Beer-Lambert se escribe como
- Las absorbancias molares desconocidas de todas las especies "coloreadas" se encuentran utilizando el método no iterativo de mínimos cuadrados lineales, una longitud de onda a la vez. Los cálculos se realizan una vez en cada ciclo de refinamiento, utilizando los valores constantes de estabilidad obtenidos en ese ciclo de refinamiento para calcular los valores de concentración de las especies en la matriz..




Errores de parámetros y correlación [ editar ]
En la región cercana al mínimo de la función objetivo, U , el sistema se aproxima a un sistema lineal de mínimos cuadrados, para el cual

Por lo tanto, los valores de los parámetros son (aproximadamente) combinaciones lineales de los valores de los datos observados y los errores en los parámetros, p , se pueden obtener por propagación de error de las observaciones, y obs , usando la fórmula lineal. Deje que la matriz de varianza-covarianza para las observaciones se denote por Σ y la de los parámetros por Σ p . Entonces,

Cuando W = ( Σ y ) −1 , esto se simplifica a

En la mayoría de los casos, los errores en las observaciones no están correlacionados, de modo que Σ y es diagonal . Si es así, cada peso debe ser el recíproco de la varianza de la observación correspondiente. Por ejemplo, en una titulación potenciométrica , el peso en un punto de titulación, k , puede ser dado por

donde σ E es el error en el potencial del electrodo o pH, ( ∂ E∂ v )
k es la pendiente de lacurva de titulaciónyσves el error en el volumen agregado.
k es la pendiente de lacurva de titulaciónyσves el error en el volumen agregado.
Cuando se utilizan pesos unitarios ( W = I , p = ( J T J ) −1 J T y ), se da a entender que los errores experimentales no están correlacionados y son iguales: Σ y = σ 2 I , donde σ 2 se conoce como varianza de una observación de peso unitario, y I es una matriz de identidad . En este caso σ 2 se aproxima por

donde U es el valor mínimo de la función objetivo y n d y n p son el número de datos y parámetros, respectivamente.

En todos los casos, la varianza del parámetro p i viene dada por Σ p
ii y la covarianza entre los parámetros p i y p j viene dada por Σ p
ij . La desviación estándar es la raíz cuadrada de la varianza. Estas estimaciones de error reflejan solo errores aleatorios en las mediciones. La verdadera incertidumbre en los parámetros es mayor debido a la presencia de errores sistemáticos , que, por definición, no se pueden cuantificar.
ii y la covarianza entre los parámetros p i y p j viene dada por Σ p
ij . La desviación estándar es la raíz cuadrada de la varianza. Estas estimaciones de error reflejan solo errores aleatorios en las mediciones. La verdadera incertidumbre en los parámetros es mayor debido a la presencia de errores sistemáticos , que, por definición, no se pueden cuantificar.
Tenga en cuenta que aunque las observaciones pueden no estar correlacionadas, los parámetros siempre están correlacionados .
Constantes derivadas [ editar ]
Cuando las constantes acumulativas se han refinado, a menudo es útil derivar constantes por pasos a partir de ellas. El procedimiento general es anotar las expresiones de definición para todas las constantes involucradas y luego igualar las concentraciones. Por ejemplo, supongamos que uno desea obtener el pKa para eliminar un protón de un ácido tribásico, LH 3 , como el ácido cítrico .
![{\ displaystyle {\ begin {alineado} {\ ce {L ^ 3 -}} + {\ ce {H + <=>}} \ {\ ce {LH ^ 2 -}} &: \ [{\ ce {LH ^ 2 -}}] = \ beta _ {11} [{\ ce {L ^ 3 -}}] [{\ ce {H +}}] \\ {\ ce {L ^ 3 -}} + {\ ce {2H + <=>}} \ {\ ce {LH2 ^ -}} &: \ [{\ ce {LH2 ^ -}}] = \ beta _ {12} [{\ ce {L ^ 3-}}] [{\ ce {H +}}] ^ {2} \\ {\ ce {L ^ 3 -}} + {\ ce {3H + <=>}} \ {\ ce {LH3}} &: \ [{\ ce {LH3}}] = \ beta _ {13} [{\ ce {L ^ 3 -}}] [{\ ce {H +}}] ^ {3} \ end {alineado}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9469a2a13baf9f2641763c2138eb0ec2a4f8f9cc)
La constante de asociación paso a paso para la formación de LH 3 está dada por
![{\ displaystyle {\ ce {{LH2 ^ {-}} + H + <=> LH3 \; \ quad \ [LH3]}} = K [{\ ce {LH2 ^ {-}}}] [{\ ce { H +}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/190cfd31265e8d89591f16358e006c3b2ecd4667)
Sustituye las expresiones de las concentraciones de LH 3 y LH -
2 en esta ecuación
2 en esta ecuación
![{\ displaystyle \ beta _ {13} [{\ ce {L ^ 3 -}}] [{\ ce {H +}}] ^ {3} = K \ beta _ {12} [{\ ce {L ^ 3 -}}] [{\ ce {H +}}] ^ {2} [{\ ce {H +}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4cd322811ee949833449526dd5951896bcd26b20)
De dónde

y como p K a = −log 10 1K, su valor viene dado por



Note la numeración inversa para pK y log β. Al calcular el error en la constante escalonada, debe tenerse en cuenta el hecho de que las constantes acumulativas están correlacionadas . Por propagación de error

y

Selección de modelo [ editar ]
Una vez que se haya completado un refinamiento, se deben verificar los resultados para verificar que el modelo elegido sea aceptable. En términos generales, un modelo es aceptable cuando los datos se ajustan a un error experimental, pero no hay un criterio único para usar para hacer el juicio. Lo siguiente debe ser considerado.
La función objetivo [ editar ]
Cuando los pesos se han derivado correctamente de las estimaciones del error experimental, el valor esperadode Un d - n p es 1. [7] Por lo tanto, es muy útil estimar los errores experimentales y derivar algunos pesos razonables de ellos ya que esto es un Indicador absoluto de la bondad del ajuste.
Cuando se usan los pesos unitarios, se implica que todas las observaciones tienen la misma varianza. Se espera que Un d - n p sea igual a esa varianza.
Errores de parámetros [ editar ]
Uno querría que los errores en las constantes de estabilidad sean aproximadamente proporcionales al error experimental. Por ejemplo, con los datos de titulación del pH, si el pH se mide con 2 decimales, los errores de log 10 β no deben ser mucho mayores que 0.01. En trabajos exploratorios donde la naturaleza de las especies presentes no se conoce de antemano, se pueden probar y comparar varios modelos químicos diferentes. Habrá modelos donde las incertidumbres en la mejor estimación de una constante de equilibrio pueden ser algo o incluso significativamente mayores que σ pH, especialmente con aquellas constantes que gobiernan la formación de especies comparativamente menores, pero la decisión de cuán grande es aceptable sigue siendo subjetiva. El proceso de decisión sobre si incluir o no equilibrios comparativamente inciertos en un modelo, y para la comparación de modelos competitivos en general, puede ser objetivo y ha sido descrito por Hamilton. [7]
Distribución de residuos [ editar ]

Como mínimo en U, el sistema puede aproximarse a uno lineal, los residuos en el caso de pesos unitarios están relacionados con las observaciones por

El simétrica , idempotente matriz J ( J T T ) -1 J es conocido en la literatura estadística como la matriz de sombrero , H . Así,

y

donde I es una matriz de identidad y M r y M y son las matrices de varianza-covarianza de los residuos y observaciones, respectivamente. Esto muestra que aunque las observaciones pueden no estar correlacionadas, los residuos siempre están correlacionados.
El diagrama de la derecha muestra el resultado de un refinamiento de las constantes de estabilidad de Ni (Gly) + , Ni (Gly) 2 y Ni (Gly) -
3 (donde GlyH = glicina ). Los valores observados se muestran como diamantes azules y las concentraciones de las especies, como porcentaje del níquel total, se superponen. Los residuos se muestran en el cuadro inferior. Los residuos no se distribuyen tan aleatoriamente como se esperaría. Esto se debe a la variación de los potenciales de unión líquida y otros efectos en las interfaces de vidrio / líquido. Esos efectos son muy lentos en comparación con la velocidad a la que se establece el equilibrio.
3 (donde GlyH = glicina ). Los valores observados se muestran como diamantes azules y las concentraciones de las especies, como porcentaje del níquel total, se superponen. Los residuos se muestran en el cuadro inferior. Los residuos no se distribuyen tan aleatoriamente como se esperaría. Esto se debe a la variación de los potenciales de unión líquida y otros efectos en las interfaces de vidrio / líquido. Esos efectos son muy lentos en comparación con la velocidad a la que se establece el equilibrio.
Restricciones físicas [ editar ]
Algunas restricciones físicas son usualmente incorporadas en los cálculos. Por ejemplo, todas las concentraciones de reactantes libres y especies deben tener valores positivos y las constantes de asociación deben tener valores positivos.
Con los datos espectrofotométricos, todos los valores de absortividad (o emisividad) molar deberían ser positivos. La mayoría de los programas de computadora no imponen esta restricción en los cálculos.
Otros modelos [ editar ]
Si el modelo no es aceptable, se debe examinar una variedad de otros modelos para encontrar el que mejor se ajuste a los datos experimentales, dentro del error experimental. La principal dificultad es con las llamadas especies menores. Estas son especies cuya concentración es tan baja que el efecto en la cantidad medida está en o por debajo del nivel de error en la medición experimental. La constante para una especie menor puede resultar imposible de determinar si no hay medios para aumentar la concentración de la especie. .
Implementaciones [ editar ]
Algunos sistemas simples son susceptibles de cálculos de hoja de cálculo. [8] Se ha publicado una gran cantidad de programas de computadora para el cálculo de la constante de equilibrio. Ver [9] para una bibliografía. Los programas más utilizados son:
- Datos potenciométricos: Hyperquad , BEST [10] PSEQUAD [11]
- Datos espectrofotométricos: HypSpec , SQUAD, [11] Specfit, [12] ReactLab EQUILIBRIA .
- RMN de datos de RMNMR , EQNMR
- Datos calorimétricos HypΔH . Los calorímetros de titulación isotérmica comercial de Affinimeter se suministran generalmente con un software con el que se puede obtener una constante de equilibrio y una entalpía de formación estándar para la formación de un aducto 1: 1. También se puede suministrar algún software para manejar equilibrios más complejos.
No hay comentarios:
Publicar un comentario