Rivaroxaban(Xarelto) fue el primer inhibidor aprobado de FXa en comercializarse en Europa y Canadá en 2008. [1] El segundo fue apixaban (Eliquis), aprobado en Europa en 2011 [2] y en Estados Unidos en 2012. [ 3] la tercera edoxabán(Lixiana, Savaysa) fue aprobado en Japón en 2011 y en Europa y en los EE.UU. en 2015. [4] betrixabán(Bevyxxa) fue aprobado en los EE.UU. en 2017.
Historia [ editar ]
Heparina [ editar ]

La heparina fue descubierta por Jay McLean y William Henry Howell en 1916, se aisló por primera vez de un hígado canino, que en griego se traduce como hepar . La heparina ataca múltiples factores en la cascada de coagulación de la sangre, uno de ellos es FXa. Al principio, tenía muchos efectos secundarios, pero durante los siguientes veinte años, los investigadores trabajaron con heparina para mejorarla y hacerla más segura. Entró en ensayos clínicos en 1935 y el primer fármaco se lanzó en 1936. Las cadenas de heparina natural pueden variar de 5.000 a 40.000 Dalton. En la década de 1980, se desarrolló heparina de bajo peso molecular (HBPM) y solo contienen cadenas con un peso molecular promedio de menos de 8.000 Da. [5]
Warfarina [ editar ]

En la década de 1920 hubo un brote de una misteriosa enfermedad hemorrágica del ganado en Canadá y el norte de los Estados Unidos. La enfermedad se denominó enfermedad del trébol dulce porque el ganado había pastado con heno de trébol dulce. No fue hasta diez años después del brote, que un investigador local, Karl P. Link y su estudiante Wilhelm Schoeffel comenzaron una investigación intensa para encontrar la sustancia que causaba la hemorragia interna . Les tomó 6 años descubrir el dicoumarol , el agente causante. [5] Ellos patentaron el derecho para la sustancia y en 1945 Link comenzó a vender un derivado de cumarina como un rodenticida . Él y sus colegas trabajaron en varias variaciones y terminaron con una sustancia que llamaron warfarina.en 1948. No fue hasta 1954 que se aprobó para uso medicinal en humanos haciendo de la warfarina el primer fármaco anticoagulante oral . [6]
Necesidad de fármacos orales más nuevos y mejores [ editar ]
El tratamiento con warfarina requiere monitoreo de sangre y ajustes de dosis regularmente debido a su estrecha ventana terapéutica . Si la supervisión no es adecuada, la warfarina representa una amenaza para causar eventos hemorrágicos demasiado frecuentes y múltiples interacciones con los alimentos y otras drogas. Actualmente, el principal problema con la heparina de bajo peso molecular (HBPM) es la vía de administración, ya que debe administrarse por vía subcutánea . [7] Debido a estas desventajas, ha habido una necesidad urgente de mejores medicamentos anticoagulantes. Para una sociedad moderna, la administración conveniente y rápida de medicamentos es la clave para un buen cumplimiento de los medicamentos . En 2008, el primer inhibidor directo de Xa fue aprobado para uso clínico. [8]Los inhibidores Direct Xa son tan eficaces como la HBPM y la warfarina, pero se administran por vía oral y no necesitan un control estricto. [7] Otras ventajas de los inhibidores de Xa son el inicio / desplazamiento rápido, pocas interacciones farmacológicas y farmacocinética predecible . El rápido efecto de inicio / desplazamiento reduce en gran medida la necesidad de "puente" con anticoagulantes parenterales después de las cirugías. [9] En la actualidad hay cuatro inhibidores del factor Xa comercializados: rivaroxabán , apixaban , Edoxabán y betrixabán . [7]
Antistasina y péptido anticoagulante de la garrapata (TAP) [ editar ]
El factor Xa fue identificado como un objetivo prometedor para el desarrollo de nuevos anticoagulantes a principios de los años ochenta. En 1987, se aisló el primer inhibidor del factor Xa, el compuesto natural de antistasina, de las glándulas salivales de la sanguijuela mexicana Haementeria officinalis . La antistasina es un polipéptido y un potente inhibidor de Xa. En 1990 se aisló otro inhibidor de Xa de origen natural, el péptido anticoagulante de garrapata (TAP) a partir de extractos de la garrapata Ornithodoros moubata . Se usaron TAP y antistasina para estimar el factor Xa como un objetivo farmacológico. [8]
Mecanismo de acción [ editar ]

La coagulación de la sangre es un proceso complejo por el cual la sangre forma coágulos. Es una parte esencial de la hemostasia y funciona al detener la pérdida de sangre de los vasos sanguíneos dañados. [10] En el sitio de la lesión, donde hay una exposición de sangre debajo del endotelio , las plaquetas se juntan y forman un tapón de inmediato. Ese proceso se llama hemostasia primaria. Simultáneamente, se produce una hemostasia secundaria. Se define como la formación de fibrina insoluble por factores de coagulación activados, específicamente la trombina. [11] Estos factores se activan entre sí en una cascada de coagulación de la sangre que se produce a través de dos vías separadas que interactúan, la vía intrínseca y la extrínseca. [12]Después de activar varias proenzimas, la trombina se forma en los últimos pasos de la cascada, luego convierte el fibrinógeno en fibrina, lo que conduce a la formación de coágulos. [10] El factor Xa es una serina proteasa activada que ocupa un papel clave en la vía de coagulación de la sangre al convertir la protrombina en trombina. La inhibición del factor Xa conduce a efectos antitrombóticos al disminuir la cantidad de trombina. Dirigirse directamente al factor Xa se sugiere como un enfoque eficaz para la anticoagulación. [8]
Desarrollo [ editar ]

En 1987 se probó la antistasina como el primer inhibidor directo de Xa. La antistasina es una proteína compuesta por 119 residuos de aminoácidos, de los cuales 20 son cisteínas involucradas en 10 enlaces disulfuro . [13] Actúa como un inhibidor lento y de unión estrecha del factor Xa con un valor de Ki de 0,3 a 0,6 nM, pero también inhibe la tripsina . [8] La antistasina recombinante puede ser producida por levaduras genéticamente modificadas, s accharomyces cerevisiae . [14]Otro inhibidor directo de Xa natural, el péptido anticoagulante de garrapata (TAP), se descubrió en 1990. Es un péptido de cadena única, 60 aminoácidos y, como la antistasina, es un inhibidor de unión estrecha y lento con un valor de Ki similar ( ~ 0.6 nM).
Estas dos proteínas se utilizaron principalmente para validar el factor Xa como un objetivo farmacológico . Los estudios en animales sugirieron que la inhibición directa de Xa era un método más eficaz para la anticoagulación en comparación con los inhibidores directos de la trombina, especialmente ofreciendo una ventana terapéuticamás amplia y reduciendo el riesgo de trombosis de rebote (aumento de eventos tromboembólicos poco después de la retirada de un medicamento antitrombótico) En comparación con los inhibidores directos e indirectos de la trombina . [8]
Durante la década de 1990, se desarrollaron varias sustancias de bajo peso molecular, como DX-9065a [15] y YM-60828. [dieciséis]
_and_DX-9065a_(right).jpg)
DX-9065a fue el primer compuesto sintético que inhibió FXa sin inhibir la trombina. Esto se logró insertando un grupo carboxilo que parecía ser el resto más importante para una unión selectiva a FXa. [8] Esas moléculas pequeñas desarrolladas tempranamente todavía tenían grupos de amidina o incluso funciones básicas superiores, que se consideraban necesarias como imitadores de un residuo de arginina en la protrombina , el sustrato natural del factor Xa. Sin embargo, estas funciones básicas también están relacionadas con una biodisponibilidad oral muy deficiente (por ejemplo, 2–3% para DX-9065a).
En 1998 , una compañía farmacéutica Bayer Healthcare comenzó a buscar inhibidores directos del factor Xa de bajo peso molecular con mayor biodisponibilidad oral. La selección de alto rendimiento y la optimización adicional al principio conducen a varias sustancias de la clase de isoindolinonas que demuestran que muchas menos sustancias básicas también pueden actuar como potentes inhibidores de Xa a un valor de IC 50 de hasta 2 nM. Aunque las isoindolinonas tienen una mejor biodisponibilidad oral que los compuestos originales, no fue suficiente. Sin embargo, el proyecto más tarde condujo a la clase de n-ariloxazolidinonas que proporciona sustancias con alta potencia de inhibición del factor Xa y alta biodisponibilidad. [8]Un compuesto de esta clase, Rivaroxabán (CI 50 = 0.7 nM, biodisponibilidad: 60%), recibió una autorización de comercialización para la prevención del tromboembolismo venoso en Europa y Canadá en septiembre de 2008. [1] [17]
Química [ editar ]
Factor Xa: Estructura y sitios de unión [ editar ]

Los factores IIa, Xa, VIIa, IXa y XIa son enzimas proteolíticas que tienen un papel específico en la cascada de coagulación. El factor Xa (FXa) es el más prometedor debido a su posición en la intersección de la vía intrínseca y extrínseca, además de generar alrededor de 1000 moléculas de trombina por cada molécula Xa, lo que resulta en un potente efecto anticoagulante. FXa se genera a partir de FX por escisión de un péptido de activación de 52 aminoácidos, ya que la "a" en el factor Xa significa activado. FXa consiste en un dominio catalítico de 254 aminoácidos y también está vinculado a una cadena ligera de 142 aminoácidos. La cadena contiene tanto el dominio GLA como dos dominios del factor de crecimiento epidérmico (dominios similares a EGF). [18]
El sitio activo de FXa está estructurado para catalizar la escisión de sustratos fisiológicos y escinde PhePheAsnProArg-ThrPhe y TyrIleAspGlyArg-IleVal en protrombina. FXa tiene cuatro llamados bolsillos que son objetivos para que los sustratos se unan al factor Xa. Estos bolsillos están alineados por diferentes aminoácidos y los inhibidores de Xa se dirigen a estos bolsillos cuando se unen al factor Xa. Los dos bolsillos más relevantes con respecto a la afinidad y selectividad para los inhibidores de Xa son S1 y S4. [18]
S1: El bolsillo S1 es un bolsillo hidrófobo y contiene un residuo de ácido aspártico (Asp-189) que puede servir como un sitio de reconocimiento para un grupo básico. FXa tiene un espacio residual en el bolsillo S1 y está recubierto por los residuos Tyr -228, Asp -189 y Ser-195. [18]
S2: El bolsillo S2 es un bolsillo pequeño y poco profundo. Se fusiona con el bolsillo S4 y tiene espacio para pequeños aminoácidos. Tyr-99 parece bloquear el acceso a este bolsillo, por lo que este bolsillo no es tan importante como S1 y S4. [19]
S3: El bolsillo S3 se encuentra en el borde del bolsillo S1 y es plano y está expuesto al solvente. Este bolsillo no es tan importante como S1 y S4.
S4: El bolsillo S4 es de naturaleza hidrófoba y el suelo del bolsillo está formado por el residuo Trp-215. Los residuos Phe-174 y Tyr-99 de FXa se unen a Trp-215 para formar una caja aromática que es capaz de unirse a fragmentos alifáticos, aromáticos y cargados positivamente. Debido a la unión a entidades cargadas positivamente, se puede describir como un agujero de cationes. [18]
Estructura química y propiedades de los inhibidores directos de Xa [ editar ]
| Rivaroxabán | Apixabán | Edoxabán | |
|---|---|---|---|
| MW (g / mol) | 436 | 460 | 548 |
| Fórmula molecular | C 19 H 18 ClN 3 O 5 S | C 25 H 25 N 5 O 4 | C 24 H 30 ClN 7 O 4 S |
| Forma | L | L | L |
| K i | 0.4 nM | 0,08 nM | 0.561 nM |
| IC 50 | 0.7 nM | N / A | N / A |
| Biodisponibilidad oral (%) | 66–100 (dependiente de la dosis) | 50 | 62 |

Unión de los inhibidores de Xa al factor Xa [ editar ]
Todos los inhibidores de Xa se unen en forma llamada forma de L dentro del sitio activo del factor Xa. Los constituyentes clave del factor Xa son los sitios de unión S1 y S4. Primero se observó que los compuestos naturales, antistasina y TAP, que poseen componentes altamente polares y, por lo tanto, cargados, se unen al objetivo con cierta especificidad. Es por eso que los medicamentos más nuevos se diseñaron con grupos con carga positiva, pero esos resultados dieron como resultado una biodisponibilidad deficiente. En la actualidad, los inhibidores de Xa comercializados, por lo tanto, contienen un anillo aromático con varios restos unidos para diferentes interacciones con los sitios de unión S1 y S4. Esto también garantiza una buena biodisponibilidad, así como el mantenimiento de una fuerza de unión firme. Los inhibidores de Xa actualmente en el mercado actual, por lo tanto, dependen de enlaces hidrófobos y de hidrógeno en lugar de interacciones altamente polares. [20]
La unión de antistasina al factor Xa [ editar ]
La antistasina contiene un dominio N y un terminal C que son similares en sus secuencias de aminoácidos con una identidad de ~ 40% y una homología de ~ 56% . Cada uno de ellos contiene una estructura de hoja β corta y 5 enlaces disulfuro . Solo el dominio N-terminal es necesario para inhibir Xa mientras que el dominio C-terminalno contribuye a las propiedades inhibitorias debido a las diferencias en la estructura tridimensional, aunque el dominio C-terminal tiene un patrón fuertemente análogo al sitio activo real . [13]
La interacción de antistasina con FXa implica tanto el sitio activo como la superficie inactiva de FXa. El sitio reactivo de la antistasina formado por Arg-34 y Val-35 en el dominio N-terminal se adapta al sitio de unión de FXa, probablemente el bolsillo S1. Al mismo tiempo, Glu-15 ubicado fuera del sitio reactivo de la antistasina se ajusta a los residuos cargados positivamente en la superficie de FXa. La unión múltiple es termodinámicamente ventajosa y conduce a una inhibición sub-nanomolar ( Ki = 0.3–0.6 nM [8] ). [13]
DX-9065a vinculante al factor Xa [ editar ]
DX-9065a, la primera molécula pequeña inhibidora de Xa directa, es un derivado de amidinoarilo con un peso molecular de 571.07 g / mol. [21] Su grupo amidinonaftaleno con carga positiva forma un puente salino hacia el residuo Asp -189 en el bolsillo S1 de FXa. El anillo de pirrolidina encaja entre Tyr-99, Phe-174 y Trp-215 en el bolsillo S4 de FXa. [22]
A diferencia de los medicamentos más antiguos, por ejemplo la heparina, el DX-9065a es selectivo para FXa en comparación con la trombina, aunque FXa y la trombina son similares en su estructura. Esto se debe a una diferencia en el residuo de aminoácido en la posición del homólogo 192. Mientras que FXa tiene un residuo de glutamina en esa posición, la trombina tiene un ácido glutámico que causa la repulsión electrostática con el grupo carboxilo de DX-9065a. Además, un puente salino entre Glu-97 de trombina y el grupo amidina fijado en el anillo de pirrolidina de DX-9065a reduce la flexibilidad de la molécula DX-9065a, que ahora no puede girar lo suficiente para evitar el choque electrostático. Es por eso que el IC 50 valor para la trombina es> 1000μM mientras que el IC 50 valor para el factor Xa es 0,16μM. [22]

Unión de rivaroxabán al factor Xa [ editar ]
La unión de rivaroxabán a FXa está mediada a través de dos enlaces de hidrógeno al aminoácido Gly-219. Estos dos enlaces de hidrógeno desempeñan un papel importante al dirigir el medicamento a los subsitios S1 y S4 de FXa. El primer enlace de hidrógeno es una interacción fuerte que proviene del oxígeno carbonilo del núcleo de oxazolidinona de rivaroxaban. El segundo enlace de hidrógeno es una interacción más débil y proviene del grupo amino del grupo clorotiofeno carboxamida.
Estos dos enlaces de hidrógeno dan como resultado que el medicamento forme una forma de L y se ajusta a los bolsillos S1 y S4. Los residuos de aminoácidos Phe-174, Tyr-99 y Trp-215 forman un estrecho canal hidrófobo que es el bolsillo de unión a S4. La parte de morfolinona de rivaroxabán está "intercalada" entre los aminoácidos Tyr-99 y Phe-174 y el anillo de arilo de rivaroxabán se orienta perpendicularmente a través de Trp-215. El grupo carbonil de la morfolinona no tiene una interacción directa con el esqueleto de FXa, en cambio, contribuye a una planarización del anillo de morfolinona y, por lo tanto, apoya que el rivaroxabán se intercale entre los dos aminoácidos.
La interacción entre el sustituyente de cloro del resto tiofeno y el anillo aromático de Tyr-228, que se encuentra en la parte inferior del S1, es muy importante debido a que elimina la necesidad de grupos fuertemente básicos para una alta afinidad por FXa. Esto permite al rivaroxaban, que no es básico, para lograr una buena biodisponibilidad y potencia oral. [8]

Apixaban vinculante al factor Xa [ editar ]
Apixaban muestra un modo de unión similar al rivaroxaban y forma un complejo inhibidor-enzima apretado cuando se conecta a FXa. El grupo p-metoxi de apixaban se conecta al bolsillo S1 de FXa, pero no parece tener ninguna interacción con ningún residuo en esta región de FXa. El átomo de nitrógeno pirazol N-2 de apixaban interactúa con Gln-192 y el oxígeno carbonilo interactúa con Gly-216. El grupo fenilactámico de apixaban se coloca entre Tyr-99 y Phe-174 y, debido a su orientación, puede interactuar con Trp-215 del bolsillo S4. El grupo carbonilo-oxígeno del grupo lactama interactúa con una molécula de agua y no parece interactuar con ningún residuo en el bolsillo S4. [23]

Estructura-actividad-relación (SAR) [ editar ]
Una parte importante del diseño de un compuesto, que es un inhibidor ideal para un determinado objetivo, es entender la secuencia de aminoácidos del sitio del objetivo al que se une el compuesto. El modelado de protrombina y FXa hace posible deducir la diferencia e identificar los aminoácidos en cada sitio de unión. En la parte inferior de la bolsa S1 en FXa, el aminoácido de unión es Asp-189, al que se pueden unir los restos de amidina. Después de realizar una radiografía del sitio de unión de FXa, se reveló que el bolsillo S1 tenía una forma plana, lo que significa que un grupo amidinoarilo plano debería unirse a él sin impedimento estérico. [8]
Los modernos inhibidores directos de Xa son moléculas en forma de L cuyos extremos encajan perfectamente en los bolsillos S1 y S4. El lado largo de la forma en L debe ajustarse a un túnel altamente específico dentro del sitio activo del objetivo. Para lograrlo, esta parte de las moléculas está diseñada para tener pocas interacciones formales con FXa en esa región. Como no hay un enlace específico, el ajuste de estos agentes entre las bolsas de FXa aumenta la especificidad total de los fármacos a la molécula de FXa. La interacción entre el bolsillo S1 de FXa y el inhibidor puede ser tanto iónica como no iónica, lo que es importante porque permite que el diseño del resto se ajuste para aumentar la biodisponibilidad oral. Los compuestos diseñados previamente eran moléculas cargadas que no se absorben bien en el tracto gastrointestinal y, por lo tanto, no alcanzan altas concentraciones séricas.[20]
Rivaroxabán
Durante el desarrollo de rivaroxaban con SAR, los investigadores se dieron cuenta de que agregar un grupo de 5-clorotiofeno-2-carboxamida al núcleo de oxazolidonina podría aumentar la potencia en 200 veces, que anteriormente había sido demasiado débil para uso médico. Además de este descubrimiento, se confirmó una clara preferencia por la configuración (S). Este compuesto tenía un perfil farmacocinético prometedor y no contenía un grupo de amidina muy básico, pero que anteriormente se había considerado importante para la interacción con el bolsillo S1. Estos hallazgos conducen a una SAR extensa (relación estructura-actividad).) investiga. Durante la prueba de SAR, R1 se definió como el grupo más importante para la potencia. La pirrolidinona fue el primer grupo funcional R1 en aumentar significativamente la potencia, pero las investigaciones adicionales revelaron una potencia aún mayor con un grupo de morfolinona. Los grupos R2 y R3 tenían hidrógeno o flúor unidos y se evaluó rápidamente que tener hidrógeno daba como resultado la mayor potencia. Los grupos R2 y R3 fueron sustituidos por varios grupos, que eran todos menos potentes que el hidrógeno, por lo que el resultado final fue hidrógeno. Como el resto clorotiofeno tenía una solubilidad en agua inadecuada, se intentó sustituirlo por otro grupo, pero no tuvo éxito. El resto clorotiofeno se une a Tyr-228 en la parte inferior de la bolsa S1, lo que lo convierte en un factor clave con respecto a la unión a FXa. Rivaroxaban tiene una alta afinidad y una buena biodisponibilidad.[24]

Apixabán

Durante el desarrollo SAR de apixaban, había tres grupos que debían ser probados para alcanzar la máxima potencia y biodisponibilidad. El primer grupo que se probó fue el sitio no activo, ya que debe estabilizarse antes de la prueba SAR en el grupo p-metoxifenilo (grupo de unión S1). Hay varios grupos que aumentan la potencia del compuesto, principalmente amidas , aminas y tetrazoles, pero también grupos metilsulfonilo y trifluorometilo. De estos grupos, la carboxamida tiene la mayor unión y tuvo una actividad de coagulación similar a la de los compuestos. [25]
En las pruebas con perros, este compuesto con un grupo de carboxamida llamado 13F, mostró un gran perfil farmacocinético, un bajo aclaramiento y una vida media y un volumen de distribución adecuados . Debido al éxito de encontrar un grupo estabilizador, se suspendió la investigación de SAR para el resto de unión S1 (p-metoxifenilo). En el grupo de unión a S4, los análogos de N-metilacetilo y lactama demostraron tener una afinidad de unión muy alta por FXa, mostraron una gran coagulación y selectividad frente a otras proteasas. La orientación resultó ser importante ya que N-metil acetilo, en comparación con la acetamida , tenía una capacidad de unión 300 veces menor a FXa debido a una planaridad desfavorable cerca del sitio de unión de la región S4. [25]
Síntesis [ editar ]
Rivaroxabán [ editar ]
El rivaroxabán químicamente pertenece al grupo de las n-ariloxazolidinonas . Otros medicamentos de ese grupo son linezolid y tedizolid , ambos antibióticos . En 2016 se publicó una síntesis de n-ariloxazolidinonas a partir de un (2,3-dihidroxipropil) -carbamato de etilo protegido con O-sililo. En una reacción en un solo recipiente, el carbamato se cicla a un anillo de 2-oxazolidona en condiciones ligeramente básicas mientras que al mismo tiempo El nitrógeno de oxazolidona es arilizado por cobre - catalización. En particular, para rivaroxaban, la 3-morfolinona sustituye el yodo en la posición p del anillo de benceno por-Catalización del cobre . Posteriormente, el grupo protector de sililo se elimina y el alcohol resultante se reemplaza por un grupo amino que luego se acilaen el último paso. [26]
Una preparación industrial de rivaroxaban fue registrado como patente por Bayer Healthcare en 2005. [27] Se inicia a partir de N- (4-aminofenol) -morpholinone que es alquilada por un óxido de propileno derivado que también contiene una amina primaria involucrado en un ftalimida protección grupo. A continuación, se agrega un equivalente de fosgeno para formar el anillo de 2-oxazolidona y se elimina la ftalimida. La amina libre ahora puede ser acilada lo que conduce a rivaroxaban.
Sin embargo, según la patente, la síntesis tiene "varias desventajas en el manejo de la reacción que tiene efectos particularmente desfavorables para la preparación". La patente también explica otra síntesis a partir de un derivado de cloro tiofeno que sería más adecuado para el proceso industrial, pero señala que los solventes o reactivos tóxicos deben eliminarse del producto final. Por lo tanto, este camino no es una alternativa. [27]
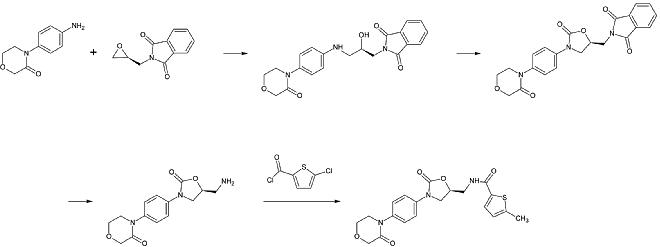
Apixaban [ editar ]
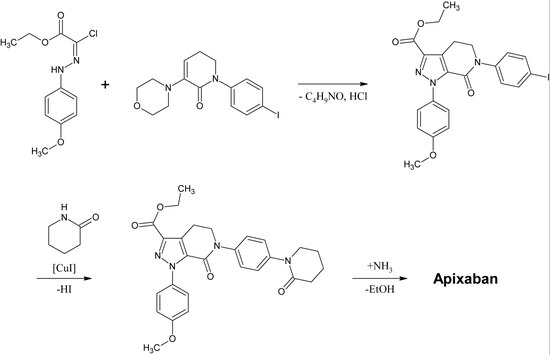
La primera síntesis completa de apixabán se publicó en 2007. [30] El paso clave de esta reacción es una cicloadición de un derivado de p-metoxifenilclorohidracón y un derivado de p-yodofenil-morfolin-dihidropiridina. Después de la siguiente eliminación de HCl y morfolina , el yodo se sustituye por 2-piperidinona por el catalizador de cobre y la éster etílico se convierte en una amida ( aminolisis ). Esta reacción se registró como patente en 2009. [31]
Uso clínico [ editar ]
Los inhibidores del factor Xa directo se utilizan clínicamente y su uso aumenta constantemente. Están tomando gradualmente el uso de warfarina y heparinas de bajo peso molecular ( HBPM ). [8] La indicación de los inhibidores de Xa es prevenir la trombosis venosa profunda (TVP) que puede conducir a una embolia pulmonar . También se usa para tratar la fibrilación auricular para reducir el riesgo de accidente cerebrovascular causado por un coágulo de sangre. Otra indicación es un tratamiento profiláctico para la coagulación de la sangre ( trombosis) debido a la aterosclerosis . Rivaroxabán fue el primer inhibidor de FXa en el mercado y luego lo siguieron apixabán, edoxabán y betrixabán .
| Rivaroxabán | Apixabán | Edoxabán | Betrixaban | |
|---|---|---|---|---|
| Nombre de la marca | Xarelto | Eliquis | Savaysa, Lixiana | Bevyxxa |
| Desarrollador y productor | Bayer | Pfizer | Daiichi Sankyo | Portola productos farmacéuticos |
Farmacocinética [ editar ]
| Rivaroxabán | Apixabán | Edoxabán | |
|---|---|---|---|
| Metabolismo | CYP3A4 / 5 (mayor), CYP2J2 (menor) | CYP3A4 (major), CYP1A2, 2C8, 2C19, 2J2 (todos menores) | CYP34A (major) |
| Enlace proteico (%) | 92–95 | 87 | 55 |
| Vida media (horas) | 5-9 | 6–12 | 5-11 |
| Eliminación | Renal (66%; 36% como fármaco sin cambios) | Renal (27%), fecal | Renal (35%) |
| Absorción (Tmax) | 2–4 horas | 3–4 horas | 1-2 horas |
| Distribución (L) | 50 | 21–61 | 107 |
| Aclaramiento renal (L / hr) | 2.4 | 7.5 | 11 |
Perspectivas de futuro [ editar ]
Inhibidores de Xa directos en los ensayos clínicos [ editar ]
Rivaroxaban, apixaban, edoxaban y betrixaban ya están en el mercado. A partir de octubre de 2016, varios nuevos inhibidores directos de Xa han entrado en ensayos clínicos. Estos son letaxaban de Takeda y eribaxaban de Pfizer. [34]
Andexxa de Portola Pharmaceuticals es una proteína recombinante que se administra por vía intravenosa . Funciona como un antídoto para todos los inhibidores directos e indirectos de FXa. Andexxa actúa como un receptor señuelo para los inhibidores de Xa.
No hay comentarios:
Publicar un comentario