Farmacocinética
En farmacología, específicamente farmacocinética, la absorción es un término que define el movimiento de un fármaco hacia el torrente sanguíneo. La absorción es el enfoque primario del desarrollo de medicinas y en la química médica, puesto que las diversas drogas deben tener la habilidad de ser absorbidas antes de que los clínicos tengan resultado. Más aún, el perfil farmacocinético de la droga puede ser cambiado con facilidad al ajustar los factores que afectan la absorción.
La absorción significa atravesar algún tipo de barrera, diferente según la vía de administración usada, pero que en último término se puede reducir al paso de barreras celulares. O dicho de otra forma, la interacción de la molécula con una membrana biológica, donde las características fisicoquímicas, tanto del fármaco como de la membrana, el resultado del proceso.
Membranas biológicas
Es indispensable conocer la estructura de la membrana citoplasmática debido a su estrecha e importante relación con la farmacocinética, que implica el pasaje de las drogas a través de las membranas. La membrana citoplasmática consiste en una capa bimolecular de lípidos, con moléculas de proteínas intercaladas, que adquiere un espesor de 75 a 80 Å (Angström, unidad de medida).1
|
|
|
|
Los fosfolípidos son responsables de las características de permeabilidad de la membrana así como eslabón importante en la cadena anabólica de numerosas sustancias de defensa (prostaglandinas, leucotrienos, ...). Suponen aproximadamente un 40% a 45% de los componentes de la membrana.
Por su parte, las proteínas constituyen alrededor del 50% de los constituyentes de las membranas, y le dan la rigidez estructural necesaria a la misma. Además, se comportan como el punto de inicio de las reacciones a las moléculas que llegan hasta la membrana (receptores), las metabolizan (enzimas), transportan moléculas en contra del gradiente de concentración a ambos lados de la membrana (bombas), o crean canales por donde puedan pasar éstas moléculas (proteínas canal).
Finalmente, nos podemos encontrar entre un 7% y un 10% de hidratos de carbono (glucolípidos y glucoproteínas) que actúan como modulador de las proteínas receptores.
EL receptor es el punto último del viaje del fármaco destinado a lograr un efecto sobre el organismo humano. De las complejas interrelaciones entre ambos se encarga otra disciplina de la farmacología: la farmacodinámica.
Vías de administración
Las barreras que ha de atravesar y las características de la absorción de cada sustancia vienen determinadas por cual haya sido la vía por la que ha llegado la misma a entrar en contacto con el organismo, o dicho de otro modo, de cual sea la vía de administración. Aquí se verá sólo una breve tabla de las diferentes vías de administración, con las características especiales en cada caso de cara a la absorción.
La vía oral es la vía recomendada para humanos. Desgraciadamente, no todos los productos pueden adaptarse para su uso por esta vía. En la vía oral el fármaco llega al organismo habitualmente después de la deglución. Una vez en el estómago, se somete a las características de los jugos del mismo, que por su acidez favorece mucho la ionización del fármaco, lo que hace que la absorción sea difícil. A pesar de todo, no son escasos los fármacos que se absorben a nivel de la mucosa gástrica: los muy liposolubles, como el alcohol o ácidos débiles como los salicilatos o los barbitúricos que presentan menores niveles de ionización a pH bajo. Cuando llega el fármaco al intestino delgado cambia el pH luminal y se favorece bastante la absorción pasiva. De hecho prácticamente todos los fármacos, menos los ácidos y bases fuertes, se absorben a este nivel. Además, en la mucosa intestinal hay numerosos mecanismos para realizar procesos de absorción en contra de gradiente, aunque difícilmente se logran niveles plasmáticos suficientes para que sean efectivos. Esta falta de absorción para algunos fármacos se aprovecha para utilizarlos a nivel local (como laneomicina o los laxantes).
La vía parenteral ofrece indudables ventajas sobre la vía oral: permite su uso en pacientes que no pueden o no deben deglutir, permite el uso de sustancias polipeptídicas y otras que se inactivan por los jugos gastrointestinales y evitan el primer paso hepático. Sin embargo precisa de instrumental para su realización y presenta inconvenientes como la infección local, tromboflebitis, neuralgias, necrosis dérmicas, etc. Desde el punto de vista farmacodinámico, la principal ventaja es la facilidad para ajustar la dosis eficaz, ya que la biodisponibilidad se considera del 100% en la mayoría de los casos.
Respecto a la vía respiratoria su interés fundamental es que brinda la posibilidad de la utilización de sustancias en estado gaseoso (casi exlusivamente oxígeno o anestésicos generales). La absorción sigue las leyes del intercambio de gases a nivel alveolar y tiene la ventaja de poner en disposición una gran superficie de absorción.
Características de la absorción
Hay que tener presente la existencia de una serie de factores que modifican la absorción:
- Solubilidad: la absorción del fármaco en más rápida cuando está en solución acuosa con respecto a si está en solución oleosa, y, a su vez, ambas son más rápidas que la que presentaría en forma sólida.
- Cinética de disolución: de la forma farmacéutica del medicamento. De la misma depende la velocidad y la magnitud de la absorción del principio activo.
- Concentración del fármaco: a mayor concentración, mayor absorción.
- Circulación en el sitio de absorción: a mayor circulación, mayor absorción.
- Superficie de absorción: a mayor superficie, mayor absorción.
Teniendo en cuenta estos factores, los mecanismos por los cuales, independientemente de la vía usada, se produce la absorción son los siguientes:
Absorción pasiva o difusión pasiva
El paso de la sustancia implicada se produce sin gasto de energía, a favor de gradientes de concentración. Puede producirse a través de la membrana propiamente dicha o a través de ciertas proteínas que forman poros.
- Difusión simple: depende del tamaño de las moléculas, y puede realizarse a través de la bicapa lipídica de la membrana o a través de los poros acuosos constituidos por las proteínas insertas en la misma. Las sustancias no ionizadas tienen mayor facilidad para la misma, siguiendo la ley de Fick, por la cual
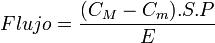
En donde C representa las concentraciones a ambos lados de la membrana, S es el área de interacción, P el coeficiente de permeabilidad y E el espesor de la membrana. Ecuación de donde se deduce que las sustancias tienden a ionizarse cuando el medio muestra un pH contrario a su naturaleza.
- Difusión facilitada: se debe a la presencia de un gradiente a ambos lados de la membrana para otras moléculas que tienen la propiedad de unirse al fármaco y arrastrarlo en su migración. Son las moléculas facilitadoras, y se incluyen dentro de la difusión pasiva debido a que no consumen energía en su trasiego. Sin embargo, a diferencia de la difusión simple, este mecanismo es saturable, al depender del número de moléculas facilitadoras.
Absorción activa o transporte activo
El paso de la sustancia implica un gasto energético en forma de moléculas de ATP. Permite la ábsorción contra gradiente y depende también de las moléculas facilitadoras, que en esta ocasión no migran en función de un gradiente, sino gracias al gasto energético. Por tanto es un mecanismo también saturable. Se realiza mediante las proteínas bomba de la membrana (ATP Binding Casete), teniendo especial transcendencia la MDR1(del inglés MultiDrug Resistence tipo 1) que exporta un gran número de fármacos y es factor clave de la resistencia de las células cancerosas a los quimioterápicos.3
La endocitosis es un mecanismo propio de algunas células por el que mediante la formación de vesículas originadas a partir de la membrana citoplásmica, introducen en su interior sustancias externas a ellas. Es un mecanismo que consume gran cantidad de energía, pero tiene la ventaja de introducir grandes cantidades de material al interior celular.
Otros factores
La absorción puede ser influenciada por factores adicionales, como la motilidad intestinal, abordajes quirúrgicos, ingesta simultánea de comida, o la presencia de otros medicamentos.
ABSORCIÓN
Velocidad con que pasa un fármaco desde su punto de administración hasta el punto de actuación.
Factores que influyen en la absorción:
ü vía de administración
ü alimentos o fluidos
ü formulación de la dosificación
ü superficie de absorción, irrigación
ü acidez del estómago
ü motilidad gastrointestinal.
Ø Vías de administración: Oral, parenteral y tópica.
1. Oral: Acción localizada (sólo hace efecto en el tubo digestivo), acción sistémica (queremos q el fármaco se absorba para q ejerza su función en otro órgano).
A esta vía también se le llama VÍA ENTERAL, por q por anatomía el fármaco, una vez q se absorbe, pasa por el hígado y posteriormente al órgano diana.
Una vez que el medicamento llega al intestino, el fármaco tiene que atravesar las barreras celulares de diferentes formas:
o Filtración: En este mecanismo los fármacos atraviesan las barreras celulares por unas soluciones de continuidad q hay en las células (canales acuosos), donde está el líquido intersticial. Por ahí pasa el fármaco por un proceso de filtración. Debe ser de bajo peso molecular para poder pasar esa barrera celular. Normalmente lo emplean sustancias q tengan carga eléctrica.
o Difusión pasiva: La utilizan los fármacos liposolubles. Se disuelven en la membrana celular, dependiendo de su liposolubilidad atraviesa la doble capa lipídica. Características:
· Fármaco liposoluble ( si no, no puede pasar)
· A favor de gradiente de concentración (de donde hay más, a donde hay menos)
· No consume energía
La liposolubilidad depende del coeficiente de partición lípido-agua, del estado de ionización.
o Difusión facilitada: El fármaco necesita un transportador para atravesar la membrana, ya que no es liposoluble. Pero si existe transportador (específico o inespecífico) se une a él y se hace liposoluble para atravesar la membrana. Una vez dentro, suelta al fármaco y sale fuera para buscar otro. Tiene las mismas características q la difusión pasiva.
o Transporte aditivo: Cuando el fármaco no liposoluble no tiene otra forma de atravesar, se une a un transportador q le ayuda a pasar la barrera celular. Lo suelta en el torrente sanguíneo. Lo q lo diferencia del anterior es q va contra gradiente, es decir, necesita energía.
La liposolubilidad de un fármaco viene determinada por el estado de ionización del mismo, es decir, cuanto más ionizado, menos liposoluble.
Si un fármaco tiene naturaleza de ácido débil, en un medio ácido, predomina la fracción no ionizada (liposoluble). Entonces un fármaco ácido débil ¿dónde se absorbe mejor? En pH ácido, por la misma regla.
Un fármaco base débil en medio básico, predomina la fracción no ionizada (liposoluble), entonces se absorbe bien en medio básico.
2. Parenteral: Podemos distinguir:
1. Vía sublingual: El medicamento se absorbe bien por los vasos de la lengua (venas raninas) que van directamente a la cava y al corazón, no pasan por el hígado. Ejemplos:Cafinitrina (tratamiento de la angina de pecho), Captopril (antihipertensivo), liotabs(Feldene flash), Alprazolan (Trankimazin, tratamiento agudo de crisis de ansiedad y pánico)
También se pueden absorber por las mucosas de la boca (como un caramelo), como por ejemplo el fentanilo.
2. Vía rectal: El medicamento se absorbe en la mucosa del recto. La administración se realiza por el esfínter anal. A veces esta vía puede comportarse como entérica, ya que se absorbe por el plexo hemorroidal y pasa al hígado. Es una vía de absorción muy rápida, pero no se sabe cuánto fármaco se absorbe. No se usa mucho. Ejemplos: Enema de corticoides para tratamiento de colitis ulcerosa, Diazepan que presenta absorción inmediata en las crisis convulsivas epilépticas y febriles.
3. Intramuscular: El medicamento se administra en un plano muscular y a través de los vasos linfáticos y los capilares se absorbe en 30 minutos. Los planos musculares adecuados son el glúteo, deltoides, cuadriceps.
Inconvenientes:
· Más de 10 ml no se deben administrar porque podemos producir necrosis ulcerosa por compresión.
· Puede producir abscesos por mala maniobra aséptica.
· Fármacos muy irritativos o muy continuos pueden formar quistes.
Consejos:
· Importante desinfectar la zona.
· Mejor con la nalga relajada.
· Aspirar, porque si metes la medicación en un capilar puede producir sobredosis.
4. Intradérmica: Su aplicación es en la dermis, para la buena administración se tiene que producir un habón en la superficie de la piel. Como ejemplo de esta vía podemos mencionar al Mantoux (prueba de la tuberculosa).
5. Intraarterial: Directamente a la luz arterial. No se utiliza mucho en terapéutica, sí en diagnóstico (contraste), estén, cateterismo.
6. Subcutánea: El medicamento se inyecta en el espacio subcutáneo y tarda en hacer efecto de 15-30 min. Ejemplo: Insulina.
7. Intravenosa: Administramos el fármaco en el sistema venoso. La velocidad (tiempo) de absorción es cero.
Inconvenientes:
Formación de abscesos, flebitis, que puede derivar en trombosis, provocando una embolia a distancia. La flebitis se forma por la utilización repetida de la vía. En caso de alergia al medicamento, si es por vía oral nos da tiempo a atajarlo porque el fármaco se absorbe poco a poco y las manifestaciones son más suaves, a medida que llegan a la sangre se hace más severa. En la vía intravenosa, la reacción alérgica puede ser más grave, más seria y provocar un shock anafiláctico en cuestión de minutos y es de aparición brusca.
Otro inconveniente es que el medicamento hay que administrarlo de forma lenta (1 ml/ min.), ya que si lo hacemos más rápido podemos provocar hipotensión brusca, q se denomina shock de velocidad. Al notar el medicamento, el organismo cree que hay una subida de tensión arterial, entonces, de forma defensiva, la baja provocando una hipotensión brusca.
Otro inconveniente es si el paciente tiene insuficiencia cardiaca, que podemos provocar sobrecarga cardiaca. Por esta vía y dependiendo del medicamento podemos provocar cambios en el equilibrio electrolítico.



La biodisponibilidad es un concepto farmacocinético que alude a la fracción y la velocidad a la cual la dosis administrada de un fármaco alcanza su diana terapéutica(canales, transportadores, receptores, que son macromoléculas proteicas), lo que implica llegar hasta el tejido sobre el que actúa. Esta cuantificación, necesaria para dar operatividad al concepto, es prácticamente imposible de hallar en el hombre. Por ello, se considera equivalente a los niveles alcanzados en la circulación sistémica del paciente. Así pues, en la práctica, la biodisponibilidad es el porcentaje de fármaco que aparece en plasma. De esta forma, la concentración plasmática del fármaco adquiere el valor máximo posible; es decir, la unidad y la biodisponibilidad se calcula para cada vía por comparación de la misma, mediante la siguiente fórmula:
![B = \frac{[ABC]_P . D_{IV}}{[ABC]_{IV} . D_P}](https://upload.wikimedia.org/math/b/f/6/bf6ee7721fa5f4c31d0eaca49851d136.png)
En donde 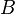 es la biodisponibilidad,
es la biodisponibilidad, 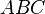 el área bajo la curva de la vía problema a determinar y de la vía intravenosa, y
el área bajo la curva de la vía problema a determinar y de la vía intravenosa, y 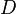 la dosis administrada en la vía intravenosa y en la vía problema a determinar.
la dosis administrada en la vía intravenosa y en la vía problema a determinar.
es la biodisponibilidad, el área bajo la curva de la vía problema a determinar y de la vía intravenosa, y la dosis administrada en la vía intravenosa y en la vía problema a determinar.
Esta determinación de la biodisponibilidad, recibe el nombre de biodisponibilidad absoluta. Sin embargo, hay fármacos que no permiten la comparación con una dosisintravenosa. En estos casos la biodisponibilidad se calcula frente a valores conocidos que se consideran estándar de otras presentaciones del mismo fármaco investigado. La fórmula quedaría entonces:
![\mathit B_R = \frac{[ABC]_{A} . dosis_{B}}{[ABC]_{B} . dosis_{A}}](https://upload.wikimedia.org/math/8/9/1/89142147383556850b8ddeefa9d174b1.png)
Es lo que se conoce como biodisponibilidad relativa ( ).
).
).
Una vez conocida la biodisponibilidad de un fármaco, podremos calcular qué modificaciones hay que realizar en su posología para alcanzar los niveles sanguíneos deseados. La biodisponibilidad es pues una razón matemática individual para cada fármaco que actúa sobre la dosis administrada. Mediante la fórmula:
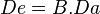
(en donde  es la dosis eficaz, la biodisponibilidad y
es la dosis eficaz, la biodisponibilidad y  la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
es la dosis eficaz, la biodisponibilidad y la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
Así, si tenemos un fármaco cuya biodisponibilidad es de 0,8 (o del 80%) y se administra una dosis de 100 mg, la ecuación se resolvería:

Este concepto depende de otra serie de factores, como son:
Forma galénica
La forma galénica utilizada tiene una importancia grande en la biodisponibilidad del fármaco. Si tomamos el ejemplo de la digoxina, mientras que el comprimido le confiere una biodisponibilidad del 70%, el elixir llega al 77% y la cápsula de gelatina o la administración intravenosa llega al 100%. El conocimiento de estas constantes nos permite hacer un cálculo adecuado cuando es preciso cambiar una vía de administración. Un ejemplo lo aclara:
Supóngase un paciente que está recibiendo comprimidos de digoxina de 0.25 mg y que por la evolución de su enfermedad presenta problemas para tragar, por lo que hay que cambiarle los comprimidos a elixir. ¿Cómo calcular la dosis correcta de elixir a administrar? Conocido lo anterior podemos aplicar la siguiente fórmula:1
 y que
y que  Por tanto,
Por tanto, 
Donde Dac, es la dosis administrada del comprimido, Bc la biodisponibilidad del comprimido, Dae la dosis administrada de elixir y Be la biodisponibilidad del elixir. Despejando,
 , es decir,
, es decir, 
Estas fórmulas no pueden aplicarse a sustancias activas que se administran como profármacos, ya que parte del fármaco puede ser eliminado antes de su activación. :3
Forma química del fármaco
Cuando el fármaco no se administra bajo su forma pura o activa, sino por ejemplo bajo la forma de una sal, hemos de conocer qué proporción del fármaco es realmente sustancia activa. Esta constante habrá que incluirla en la fórmula anterior.

donde Q es la constante de pureza del fármaco.1
Vía de administración
Los obstáculos que pone el organismo a permitir la llegada de sustancias extrañas a su interior son diferentes en función del tejido que se valore. Evidentemente, la absorción del fármaco no es igual si se da por vía oral, vía rectal, vía tópica o vía intravenosa. La biodisponibilidad de un fármaco habrá que calcularla de forma individual para cada vía en que pueda utilizarse el mismo. Sin embargo, y sobre todo en el caso de la administración intravenosa, cuya biodisponibilidad se considera axiomáticamente de valor 1, hay un nuevo factor a incluir: la velocidad de administración, que es la velocidad a la cual el fármaco absorbido alcanza la circulación sistémica. Este concepto se incorpora a la fórmula anterior mediante una constante: ( ) La fórmula quedaría:1
) La fórmula quedaría:1
) La fórmula quedaría:1 Como
Como  y si asumimos que
y si asumimos que  , entonces
, entonces 
En este punto es necesario aclarar que se entiende por circulación sistémica a aquella que va desde el corazón a los tejidos. Esto es importante tenerlo en cuenta ya que cuando se administra un fármaco por vía oral, al absorberse en el intestino viaja a través de la vena porta hasta el hígado, donde puede sufrir un primer proceso de metabolización y de aquí hasta el corazón. Evidentemente este proceso presenta una velocidad de administración más lenta que la administración directamente en vena del fármaco, la cual será a su vez más lenta que la administración intraarterial.
Estabilidad del fármaco
Los fármacos son sustancias químicas con sus propiedades inherentes. Así, podemos encontrar sustancias de naturaleza ácida o básica y dentro de ellas ácidos fuertes o débiles y bases fuertes o débiles. La diferencia es que tanto ácidos como bases débiles tienen una alta capacidad de ionización. Esto es importante debido a que las formas no ionizadas suelen ser más liposolubles y por tanto tendrán capacidad de atravesar las membranas celulares, mecanismo éste indispensable tanto para la absorción como para la excreción. Por el contrario, las formas ionizadas suelen ser más hidrosolubles, y por tanto presentan alta dificultad para atravesar las membranas celulares. No obstante, hay una parte de la sustancia que a pesar de no estar ionizada no consigue atravesar las membranas, mientras que también hay una parte que estando ionizada sí consigue atravesarlas.
El pKa de una sustancia es el pH al que una sustancia tiene la mitad de sus moléculas ionizadas y la otra mitad sin ionizar. Es una situación estable que nos da información sobre la capacidad del fármaco para atravesar de forma pasiva las membranas. La fórmula para calcular el pKa (conocida como ecuación de Henderson-Hasselbach) incluye el pH del medio, ya que en un medio ácido las bases débiles se encuentran muy ionizadas mientras que en un medio alcalino son los ácidos débiles los que se encontrarán ionizados. Fisiopatológicamente esto es importante, ya que podemos modificar, dentro de ciertos márgenes, el pH ambiental para favorecer la absorción de una fármaco (por ejemplo, tomarlo en ayunas o tras la comida) o la eliminación del mismo (alcalinizando o acidificando la orina).

Metabolización
El grado de metabolización que sufre el fármaco antes de alcanzar la circulación sistémica es otro de los factores a tener en cuenta para la valoración de la biodisponibilidad. Hay fármacos que sufren un alto índice de metabolización en su primer paso por el hígado (metabolismo de primer paso) de tal manera que incluso alguno que teóricamente puede utilizarse por vía oral, se descarta su uso debido a la poca disponibilidad alcanzada después del metabolismo de primer paso (lidocaína).
La biodisponibilidad de un fármaco es un término farmacocinético que alude a la fracción de la dosis del mismo administrada que alcanza su diana terapéutica o lo que es lo mismo que llega hasta el tejido sobre el que realiza su actividad.
En seres humanos la cuestión es un tanto más complicada, porque conocer la cantidad exacta de fármaco que llega a su diana terapéutica conllevaría tomar muestras del tejido donde se encuentre y lamentablemente extraer según qué tejidos es altamente delicado. Por eso, se toma como valor aproximado la concentración plasmática de dicho fármaco y se compara con la concentración plasmática que alcanza para ese mismo preparado una inyección intravenosa. Este cociente es al que llamamos biodisponibilidad -más concretamente biodisponibilidad absoluta-.

Gráfico descriptivo sobre la biodisponibilidad, la Concentración Mínima Eficaz y Mínima Tóxica (CME y CMT respectivamente).
BIODISPONIBILIDAD
DEFINICIONES Y CONCEPTO
La biodisponibilidad de un medicamento es un concepto relativamente nuevo de la calidad de éste. Ha ido cobrando importancia en las dos últimas décadas tras haberse demostrado en innumerables trabajos que formas farmacéuticas sólidas de administración oral (comprimidos, cápsulas, etc. ), si bien eran equivalentes desde el punto de vista de la cantidad de fármaco, no lo eran desde el punto de vista fisiológico.
Las definiciones de biodisponibilidad son numerosas:
"Un término empleado para indicar una medida de la cantidad relativa de un fármaco que llega a la circulación general y la velocidad a la cual esto ocurre".(48)
"Significa la cantidad y velocidad a la cual el principio activo es absorbido desde un producto farmacéutico y que queda disponible en el sitio de acción".(49)
"La extensión y velocidad de la absorción desde una forma farmacéutica, reflejada por la curva de concentración-tiempo del fármaco administrado, en la circulación sistémica".(2)
Todas estas definiciones coinciden en que el concepto de biodisponibilidad abarca dos componentes: la cantidad de fármaco absorbido y la velocidad de absorción.
La Administración de Alimentos y Fármacos de los Estados Unidos de América (Food and Drug Administration, FDA) ha definido además otros conceptos de uso general: (49)
"Equivalentes farmacéuticos": formas farmacéuticas que contienen idénticas cantidades del mismo principio activo, por ejemplo: la misma sal o éster, en idéntica forma farmacéutica, pero que no contienen necesariamente el mismo ingrediente inactivo (excipiente) y que cumplen con los requisitos establecidos en las farmacopeas en cuanto, a identidad, potencia, calidad y pureza y, si es aplicable, uniformidad de contenido y tiempo de desintegración y/o disolución.
"Alternativas farmacéuticas": formas farmacéuticas que contienen idéntica porción activa de la molécula o su precursor, pero no necesariamente en la misma cantidad o forma farmacéutica o la misma sal o éster. Tales formas farmacéuticas cumplen, en forma individual, con los requisitos de farmacopea en cuanto a identidad, pureza, potencia y, si es aplicable, uniformidad de contenido, desintegración y/o disolución.
"Productos bioequivalentes": equivalentes farmacéuticos o alternativas farmacéuticas cuya velocidad y extensión de la absorción no exhiben diferencias significativas cuando se administran en la misma dosis de la porción terapéutica bajo condiciones experimentales similares, ya sea en dosis única o en dosis múltiples.
Por consiguiente, la bioequivalencia implica la comparación de dos o más productos basada en la velocidad y magnitud de la absorción y, mientras esto no se verifique, dos productos no podrán ser considerados bioequivalentes aun cuando se obtengan resultados equivalentes de los ensayos tradicionales de control de calidad, de contenido y de desintegración y/o disolución.
Numerosos investigadores han demostrado que la biodisponibilidad de los productos farmacéuticos puede ser muy variable. El problema consiste en determinar si la variación de la absorción de los productos puede tener una consecuencia terapéutica debido a la producción de síntomas tóxicos o a la reducción del efecto terapéutico. En varias circunstancias es difícil visualizar una diferencia terapéutica a simple vista, especialmente en el caso de fármacos en que la curva de dosis-respuesta está relativamente enmarcada en amplios rangos de efectividad, en los cuales sólo muy pronunciadas variaciones de la biodisponibilidad pueden traducirse en respuestas terapéuticas inadecuadas, como se ha comprobado en el caso de cloranfenicol.(50)
Las características de liberación del fármaco desde la forma farmacéutica- pueden influir notoriamente en la rapidez y magnitud de la absorción, lo cual puede esquematizarse de la siguiente manera:

El proceso de disolución tiene, pues, enorme importancia en la absorción de los fármacos, ya que la mayoría de éstos son absorbidos por difusión pasiva de las moléculas disueltas. De ahí que los estudios de velocidad de disolución pueden ser de gran utilidad en la evaluación in vitro de los productos medicamentosos.(51) Por lo tanto, el principal factor en el defecto de absorción se debe, generalmente, a la deficiente liberación del principio activo, la que a su vez es causada por una deficiente formulación o la aplicación inadecuada de tecnología en la obtención de la forma farmacéutica.
No hay comentarios:
Publicar un comentario