Mutaciones
La heteroplasmia es la presencia de ADN mitocondrial de distintos tipos en la misma célula.
Se dice de un individuo que es heteroplasmático cuando presenta una mezcla de dos poblaciones diferentes de mitocondrias. Si, por el contrario, todas las mitocondrias tienen el mismo genoma, se dice que es homoplasmático.
El fenómeno de la heteroplasmia se da por acumulación de errores durante lareplicación y por los daños causados por los radicales del medio oxidante en que se encuentra la mitocondria. Los tejidos con mayor dependencia del metabolismo de la mitocondria, es decir, aquéllos con mayor demanda energética, son más sensibles a las mutaciones mitocondriales porque la replicación mitocondrial más activa se da en ellos. Si afecta al ovocito, la heteroplasmia puede transmitirse a la descendencia, y, por fenómenos de deriva o de selección, puede causar sustituciones de poblaciones.
La heteroplasmia del ADN mitocondrial está relacionada con las enfermedades mitocondriales. Es importante destacar que la detección de muchas enfermedades mitocondriales heteroplasmáticas debe hacerse a partir de biopsias de músculo esquelético o del órgano o el tejido afectado si es posible, ya que muchas de ellas no son detectables en sangre. Las técnicas más empleadas para detectar este tipo de enfermedades son:
- Southern Blot, para detectar grandes deleciones o duplicaciones.
- PCR - RFLP, para análisis de mutaciones concretas.
- Secuenciación directa del genoma mitocondrial
Con frecuencia, los síntomas de enfermedades mitocondriales severas no se manifiestan hasta la edad adulta, porque se necesitan muchas divisiones celulares y mucho tiempo para que una célula reciba la cantidad suficiente de mitocondrias con los alelos mutantes que causan la enfermedad. Un ejemplo de este fenómeno es la Atrofia Óptica de Leber: los individuos afectados no experimentan dificultades ópticas hasta que alcanzan la adultez; en otros casos, el grado de heteroplasmia explica la variación en la severidad de la enfermedad entre los descendientes.
Algunas enfermedades mitocondriales heteroplásmicas son
- Atrofia óptica de Leber.
- Miopatía mitocondrial; MELAS: encefalomiopatía mitocondrial, acidosis láctica, infartos.
- Epilepsia mioclónica (MERF).
- Síndrome de Kearns-Sayre.
- Síndrome de Pearson.
- Alta tasa de mutación: La tasa de mutación espontánea del ADNmt es unas 10 veces superior a la del ADN nuclear. Por tanto, hay una gran variación de secuencias entre especies e incluso entre individuos de una misma especie. En el hombre se ha calculado que dos individuos escogidos al azar tienen como promedio 50-70 nucleótidos diferentes en su genoma mitocondrial. Además de estas diferencias, en un individuo determinado se está produciendo continuamente, a lo largo de la vida, una heterogeneidad en el ADNmt como consecuencia de las mutaciones que se están dando en las células somáticas. Se ha propuesto que una acumulación de este daño mitocondrial pudiera ser la causa de la disminución en la capacidad respiratoria de los tejidos que tiene lugar durante el envejecimiento.
- Poliplasmia y Segregación mitótica: En cada célula hay cientos o miles de moléculas de ADNmt. En principio, todas las células de un individuo normal tienen moléculas idénticas de ADNmt, situación que se denomina homoplasmia. Si en una célula aparecen dos poblaciones de ADNmt, una normal y otra mutada, se dice que estamos en una situación de heteroplasmia. Durante la división celular, las mitocondrias —y, por tanto, las moléculas de ADNmt— se distribuyen al azar entre las células hijas.
- Efecto umbral: El fenotipo de una célula en heteroplasmia dependerá del porcentaje de ADN dañado que exista en la célula; es decir, del grado de heteroplasmia de una mutación. Cuando el número de moléculas de ADNmt mutadas es pequeño, el ADNmt normal es capaz de mantener la función mitocondrial. Sin embargo, cuando el número de copias de ADNmt mutado sobrepasa un umbral determinado, la producción de ATP puede llegar a estar por debajo de los mínimos necesarios para el funcionamiento de los tejidos, llevando al desarrollo de patología mitocondrial. Es importante tener en cuenta que los distintos tejidos pueden variar en el grado de homoplasmia o heteroplasmia para un ADNmt mutado, lo que origina una gran variabilidad en la expresividad clínica de las enfermedades mitocondriales. Por otra parte, los tejidos tienen distintas necesidades energéticas, y el número de orgánulos y de moléculas de ADNmt es también diferente en cada tejido. Por tanto, los órganos preferentemente afectados en las enfermedades mitocondriales son aquellos que dependen en mayor grado de la producción de energía para su correcto funcionamiento.
- Herencia materna: El ADNmt se hereda exclusivamente por vía materna. La pequeña cantidad de genoma mitocondrial contenido en el espermatozoide raramente entra en el óvulo fecundado, y cuando lo hace es eliminado activamente. El tipo de herencia matrilineal es bastante fácil de reconocer al examinar un árbol genealógico, y tiene importantes consecuencias en el consejo genético (ver más adelante).
Por lo que respecta a las enfermedades humanas debidas a mutaciones del ADNmt, podemos dividirlas en dos grandes grupos: enfermedades asociadas a mutaciones puntuales y enfermedades debidas a alteraciones estructurales del ADNmt. Las enfermedades asociadas a mutaciones puntuales son frecuentes, debido a la alta tasa de mutación del ADNmt. Actualmente se han descrito más de 50 mutaciones puntuales, que se localizan en los tres tipos de genes codificados en el ADNmt (ARNr, ARNt y polipéptidos del OXPHOS). Según su efecto patogénico, distinguimos las mutaciones que originan el cambio de un aminoácido por otro, afectando a alguno de los 13 genes codificantes de proteínas, y las mutaciones que afectan a losgenes de los ARNr ó ARNt, que tienen efectos globales en la síntesis de las proteínas mitocondriales. Las enfermedades asociadas a alteraciones estructurales del ADNmt pueden ser tanto deleciones más o menos grandes como, menos frecuentemente, inserciones y/o duplicaciones. Hasta el momento se han descrito varios cientos de reordenaciones del ADNmt, que —al contrario que las mutaciones puntuales— suelen ser esporádicas. Como regla práctica, podemos decir que en aquellos casos que muestran herencia matrilineal ha de sospecharse la presencia de mutaciones puntuales del ADNmt; en casos esporádicos, en cambio, son más frecuentes las deleciones y/o duplicaciones. Si, por el contrario, se confirma un patrón de herencia autosómico dominante, el estudio debe ir dirigido hacia la búsqueda de deleciones múltiples; si fuese autosómico recesivo, deben buscarse depleciones del ADNmt.
La siguiente tabla muestra algunas de las mutaciones puntuales más frecuentes en el ADN mitocondrial, incluyendo también una pequeña deleción. Se indican, de izquierda a derecha, el tipo de mutación, el cambio de aminoácido (cuando existe) y el gen afectado por la mutación. Son especialmente frecuentes las mutaciones que afectan a los nucleótidos 11.778 y 14.484 (Neuropatía óptica hereditaria de Leber), al 8.993 (Síndrome de Leigh), al 3.243 (MELAS) y al 8.344 (MERRF).
La detección de mutaciones puntuales se realiza habitualmente por digestión de fragmentos de PCR con enzimas de restricción sensibles al cambio de nucleótido introducido por la mutación. En cualquier caso, resulta más fiable la detección mediante Southern del ADNmt digerido con enzimas de restricción específicas para cada una de las mutaciones concretas, pero esto no siempre es posible y, por otra parte, es más laborioso.
Las deleciones se encuentran siempre en heteroplasmia, aunque pueden llegar a constituir un porcentaje muy alto de la población de ADNmt. Su determinación se hace fundamentalmente en biopsias musculares, pero si la afectación es muy grave se pueden llegar a detectar también en sangre periférica. Las deleciones se detectan con relativa facilidad por la técnica de Southern, que muestra poblaciones de ADNmt de menor tamaño que el normal (16.569 pb). Se emplea habitualmente ADNmt digerido con BamHI o con EcoRV, utilizando como sonda un gran fragmento de ADNmt obtenido mediante PCR de largo alcance.
El diagnóstico de depleción del ADNmt se realiza también por hibridación Southern, comparando los niveles de ADNmt con los de ADN nuclear.
La confirmación molecular de una alteración del ADNmt ayuda a establecer el diagnóstico de enfermedad mitocondrial y el patrón de herencia matrilineal. Esto tiene una importancia capital desde el punto de vista del consejo genético y el cálculo del riesgo de recurrencia de la enfermedad, ya que la herencia matrilineal está caracterizada por la transmisión exclusivamente por vía materna. Esto hace que tenga algunas características especiales: se afectan ambos sexos, pero los varones -enfermos o no- nunca transmiten la enfermedad, y sus descendientes (hijos o hijas) no son portadores; por otro lado, las mujeres afectadas transmiten la enfermedad a toda su descendencia, de forma que todas sus hijas tienen riesgo de transmitir y/o padecer la enfermedad, y todos sus hijos tienen riesgo de padecerla.

La Figura 11.15 muestra un árbol genealógico típico de herencia matrilineal. La figura muestra el pedigrí de una familia en la que se transmite una enfermedad mitocondrial. Se observa un patrón de herencia matrilineal, con transmisión siempre por vía materna: ningún varón afectado transmite la enfermedad. Además, se pueden identificar cuatro mujeres portadoras asintomáticas (flechas rojas), ya que —debido a los distintos grados de heteroplasmia— con frecuencia las mujeres que transmiten la enfermedad no lleguan a desarrollar el cuadro clínico completo.
Las enfermedades mitocondriales pueden deberse a defectos en genes nucleares o a alteraciones del propio genoma mitocondrial. La diferencia entre ambos tipos de procesos es importante porque los tipos de herencia son diferentes y las implicaciones para el cálculo de riesgos pueden ser grandes. Dada la importancia del metabolismo oxidativo, las enfermedades mitocondriales presentan gran variedad de síntomas y signos, y suponen un problema diagnóstico importante. En general encontramos afectación neuromuscular: neuropatía óptica, convulsiones, accidentes cerebrovasculares, mioclonías, neuropatía periférica, ataxia, demencia, miopatías. De todas formas, los pacientes con enfermedad del ADN mitocondrial pueden agruparse en tres categorías:
- Raramente se presenta como un síndrome fácilmente identificable: MELAS(encefalopatía mitocondrial con acidosis láctica y episodios de accidentes cerebrovasculares) muestra estatura baja, sordera bilateral, diabetes, convulsiones, accidentes cerebrovasculares y encefalopatía en la 3ª o 4ª décadas de la vida. LHON(Leber hereditary optic neuropathy) muestran fallo visual bilateral en la 2ª-3ª décadas. Otras presentaciones clásicas son la oftalmoplegia externa con o sin ptosis, que aparece junto con una miopatía proximal en CPEO (chronic progressvie external ophtalmoplegia) o con ataxia, sordera bilateral y defectos de conducción cardíaca en el Síndrome de Kearns-Sayre. Otros síndromes son NARP (neurogenic weakness, ataxia, retinitis pigmentosa) y MERRF (epilepsia mioclónica y ragged-red-fibers o fibras musculares rojas deshilachadas).
- Un segundo grupo de pacientes tienen una constelación de datos clínicos que sugieren enfermedad mitocondrial, pero no encajan en ninguno de los síndromes citados. Por ejemplo, son típicos los casos de estatura baja con sordera neurosensorial bilateral, oftalmoplegia con ptosis, diabetes y migrañas. Es necesario un estudio neurológico exhaustivo, y la asociación de cualquiera de estos datos con miopatía o signos de afectación neurológica central hace el diagnóstico de enfermedad mitocondrial muy probable. Sin embargo, lo más habitual es que estos pacientes sean estudiados para descartar gran cantidad de enfermedades neurológicas hereditarias autosómicas (alguna forma de ataxia espinocerebelosa, estadíos iniciales de la enfermedad de Huntington, síndrome de Usher, Charcot-Marie-Tooth) antes de que se llegue a pensar en enfermedad mitocondrial. Un diagnóstico común en estos pacientes es el de “myastenia congénita”.
- El último gran grupo de enfermos es el más difícil de definir, y está constituído por pacientes con síntomas aislados que -después de un estudio exhaustivo- terminan achacándose a patología mitocondrial. Por ejemplo, un cierto porcentaje de accidentes cerebrovasculares juveniles son resultado de patología mitocondrial, pero además la presentación a menudo es extra-neurológica: cardiomiopatía hipertrófica, enfermedad tubular renal, hipoparatiroidismo, insuficiencia suprarrenal, diabetes, disfagia. La sordera familiar neurosensorial progresiva es frecuentemente debida a patología mitocondrial, como demostró el grupo de Xavier Estivill en 70 familias españolas con la mutación del nucleótido 1555.
Entre las pruebas diagnósticas que resultan útiles para llegar al diagnóstico de patología mitocondrial, contamos con las siguientes:
- Investigaciones generales: habitualmente, estos pacientes han sido sometidos a gran catidad de tests. Es necesario ver la función cardíaca, test de tolerancia a glucosa, nivel de lactato en sangre (es más específico en el líquido cefalo-raquídeo), electroencefalograma para detectar encefalopatía subaguda, TAC cerebral (es común la calcificación bilateral de ganglios basales).
- Investigaciones específicas: detección de alteraciones en el ADN mitocondrial, tanto reordenamientos (deleciones, duplicaciones) como mutaciones puntuales. Lo más habitual es que las mutaciones estén en heteroplasmia, y esto determina la expresividad fenotípica (habitualmente es necesario >85% de ADNmt mutado para que se manifieste la enfermedad). Las mutaciones letales sólo pueden estar en heteroplasmia, pero en general el porcentaje de heteroplasmia varía en distintos órganos en un mismo individuo e incluso también con la edad, de ahí la enorme variabilidad clínica. En algunas entidades es relativamente sencilla la detección molecular: por ejemplo, hay 3 mutaciones que en conjunto explican el 95% de los pacientes con LHON. En cambio, las deleciones responsables de los síndromes de CPEO o Kearns-Sayre no son detectables en sangre, por lo que los resultados negativos han de ser interpretados con cautela. En estos casos, la biopsia de músculo esquéletico es la piedra angular para el diagnóstico de patología mitocondrial: por un lado, la detección histoquímica de fibras deficientes en COX y en otros complejos de la cadena respiratoria confirma la afectación mitocondrial; además, los estudios moleculares en ADN muscular confirman la presencia de deleciones o de mutaciones puntuales.
Una mutación es un cambio en la información genética (genotipo) de un ser vivo, que produce una variación en las características de este y que puede trasmitirse a su descendencia. Se presenta de manera espontánea y súbita o por la acción demutágenos. Este cambio estará presente en una pequeña proporción de la población (variante) o del organismo (mutación). La unidad genética capaz de mutar es el gen, la unidad de información hereditaria que forma parte del ADN.
En los seres pluricelulares, las mutaciones solo pueden ser heredadas cuando afectan a las células reproductivas. Una consecuencia de las mutaciones puede ser, por ejemplo, una enfermedad genética. Sin embargo, aunque a corto plazo pueden parecer perjudiciales, las mutaciones son esenciales para nuestra existencia a largo plazo. Sin mutación no habría cambio, y sin cambio la vida no podría evolucionar.
Definición
La definición que en su obra de 1901 "La teoría de la mutación" Hugo de Vries dio de la mutación (del latín mutare = cambiar) era la de cualquier cambio heredable en el material hereditario que no se puede explicar mediante segregación o recombinación. Más tarde se descubrió que lo que De Vries llamó mutación en realidad eran más bien recombinaciones entre genes.
La definición de mutación a partir del conocimiento de que el material hereditario es el ADN y de la propuesta de la doble hélice para explicar la estructura del material hereditario (Watson y Crick,1953), sería que una mutación es cualquier cambio en la secuencia de nucleótidos del ADN. Cuando dicha mutación afecta a un solo gen, se denomina mutación génica. Cuando es la estructura de uno o varios cromosomas lo que se ve afectado, mutación cromosómica. Y cuando una o varias mutaciones provocan alteraciones en todo el genoma se denominan, mutaciones genómicas.
Mutación somática y mutación en la línea germinal
- Mutación somática: es la que afecta a las células somáticas del individuo. Como consecuencia aparecen individuos mosaico que poseen dos líneas celulares diferentes con distinto genotipo. Una vez que una célula sufre una mutación, todas las células que derivan de ella por divisiones mitóticas heredarán la mutación (herencia celular). Un individuo mosaico originado por una mutación somática posee un grupo de células con un genotipo diferente al resto, cuanto antes se haya dado la mutación en el desarrollo del individuo mayor será la proporción de células con distinto genotipo. En el supuesto de que la mutación se hubiera dado después de la primera división del cigoto (en estado de dos células), la mitad de las células del individuo adulto tendrían un genotipo y la otra mitad otro distinto. Las mutaciones que afectan solamente a las células de la línea somática no se transmiten a la siguiente generación.2 3
- Mutaciones en la línea germinal: son las que afectan a las células productoras de gametos apareciendo, de este modo, gametos con mutaciones. Estas mutaciones se transmiten a la siguiente generación y tienen una mayor importancia en la evolución biológica.2 3
Tipos de mutación según sus consecuencias
Las consecuencias fenotípicas de las mutaciones son muy variadas, desde grandes cambios hasta pequeñas diferencias tan sutiles que es necesario emplear técnicas muy desarrolladas para su detección.2 3
Mutaciones morfológicas
Afectan a la morfología del individuo, a su distribución corporal. Modifican el color o la forma de cualquier órgano de un animal o de una planta. Suelen producirmalformaciones. Un ejemplo de una mutación que produce malformaciones en humanos es aquella que determina la neurofibromatosis. Esta es una enfermedad hereditaria, relativamente frecuente (1 en 3.000 individuos), producida por una mutación en el cromosoma 17 y que tiene una penetrancia del 100 % y expresividadvariable. Sus manifestaciones principales son la presencia de neurofibromas, glioma del nervio óptico, manchas cutáneas de color café con leche, hamartomas del iris, alteraciones óseas (displasia del esfenoide, adelgazamiento de la cortical de huesos largos). Con frecuencia hay retardo mental y macrocefalia.4
Mutaciones letales y deletéreas
Son las que afectan la supervivencia de los individuos, ocasionándoles la muerte antes de alcanzar la madurez sexual. Cuando la mutación no produce la muerte, sino una disminución de la capacidad del individuo para sobrevivir y/o reproducirse, se dice que la mutación es deletérea. Este tipo de mutaciones suelen producirse por cambios inesperados en genes que son esenciales o imprescindibles para la supervivencia del individuo. En general las mutaciones letales son recesivas, es decir, se manifiestan solamente en homocigosis o bien, en hemicigosis para aquellos genes ligados al cromosoma X en humanos, por ejemplo.2 5
Mutaciones condicionales
Las mutaciones condicionales (incluidas las condicionalmente letales) son muy útiles para estudiar aquellos genes esenciales para la bacteria. En estos mutantes hay que distinguir dos tipos de condiciones:
condiciones restrictivas (también llamadas no-permisivas): son aquellas condiciones ambientales bajo las cuales el individuo pierde la viabilidad, o su fenotipo se ve alterado, debido a que el producto afectado por la mutación pierde su actividad biológica.
condiciones permisivas: son aquellas bajo las cuales el producto del gen mutado es aún funcional.
Mutaciones bioquímicas o nutritivas
Son los cambios que generan una pérdida o un cambio de alguna función bioquímica como, por ejemplo, la actividad de una determinada enzima. Se detectan ya que el organismo que presenta esta mutación no puede crecer o proliferar en un medio de cultivo por ejemplo, a no ser que se le suministre un compuesto determinado. Los microorganismos constituyen un material de elección para estudiar este tipo de mutaciones ya que las cepas silvestres solo necesitan para crecer un medio compuesto por sales inorgánicas y una fuente de energía como la glucosa. Ese tipo de medio se denomina mínimo y las cepas que crecen en él se dicen prototróficas. Cualquier cepa mutante para un gen que produce una enzima perteneciente a una vía metabólica determinada, requerirá que se suplemente el medio de cultivo mínimo con el producto final de la vía o ruta metabólica que se encuentra alterada. Esa cepa se llama auxotrófica y presenta una mutación bioquímica o nutritiva.6
Mutaciones de pérdida de función
Las mutaciones suelen determinar que la función del gen en cuestión no se pueda llevar a cabo correctamente, por lo que desaparece alguna función del organismo que la presenta. Este tipo de mutaciones, las que suelen ser recesivas, se denominan mutaciones de pérdida de función. Un ejemplo es la mutación del gen hTPH2 que produce la enzima triptófano hidroxilasa en humanos. Esta enzima está involucrada en la producción de serotonina en el cerebro. Una mutación (G1463A) de hTPH2determina aproximadamente un 80 % de pérdida de función de la enzima, lo que se traduce en una disminución en la producción de serotonina y se manifiesta en un tipo de depresión llamada depresión unipolar.7
Mutaciones de ganancia de función
Cuando ocurre un cambio en el ADN, lo más normal es que corrompa algún proceso normal del ser vivo. Sin embargo, existen raras ocasiones donde una mutación puede producir una nueva función al gen, generando un fenotipo nuevo. Si ese gen mantiene la función original, o si se trata de un gen duplicado, puede dar lugar a un primer paso en la evolución. Un caso es la resistencia a antibióticos desarrollada por algunas bacterias (por eso no es recomendable hacer un uso abusivo de algunos antibióticos ya que finalmente el organismo patógeno irá evolucionando y el antibiótico no le hará ningún efecto).
Tipos de mutación según el mecanismo causal
Según el mecanismo que ha provocado el cambio en el material genético, se suele hablar de tres tipos de mutaciones: mutaciones cariotípicas o genómicas,mutaciones cromosómicas y mutaciones génicas o moleculares. En el siguiente cuadro se describen los diferentes tipos de mutaciones y los mecanismos causales de cada una de ellas.2 3
| Transición | |||||||||||||||||||||||||||||||
| Por sustitución de bases | |||||||||||||||||||||||||||||||
| Transversión | |||||||||||||||||||||||||||||||
| génica o molecular | |||||||||||||||||||||||||||||||
| INDELs (por inserciones o deleciones de bases) | |||||||||||||||||||||||||||||||
| Inversiones | |||||||||||||||||||||||||||||||
| Mutación | cromosómica | Deleciones o duplicaciones | |||||||||||||||||||||||||||||
| Translocaciones | |||||||||||||||||||||||||||||||
| Poliploidía | |||||||||||||||||||||||||||||||
| genómica | |||||||||||||||||||||||||||||||
| Aneuploidía | |||||||||||||||||||||||||||||||
Hay una tendencia actual a considerar como mutaciones en sentido estricto solamente las génicas, mientras que los otros tipos entrarían en el término de aberraciones cromosómicas.


.jpg)
Mutaciones cromosómicas

Definición
Las mutaciones cromosómicas son modificaciones en el número total de cromosomas, la duplicación o supresión de genes o de segmentos de un cromosoma y la reordenación del material genético dentro o entre cromosomas. Pueden ser vistas al microscopio, sometiendo a los cromosomas a la “técnica de bandas”. De esta manera se podrá confeccionar el cariotipo.
Introducción
- Las alteraciones de la dotación diploide de cromosomas se denominan aberraciones cromosómicas o mutaciones cromosómicas.
- Hay 3 tipos de mutaciones cromosómicas:
- Reordenamientos cromosómicos: implican cambios en la estructura de los cromosomas (duplicación, deleción,inversión , traslocación y formación de cromosomas en anillo).
- Aneuploidías:supone un aumento o disminución en el número de cromosomas.
- Poliploidia: presencia de conjuntos adicionales de cromosomas.
- La aneuploidia: da lugar a monosomías, trisomías, tetrasomías, etc.
- La poliploidia: dotaciones de cromosomas pueden tener orígenes idénticos o distintos, dando lugar a autopoliploides yalopoloploides, respectivamente.
- Las deleciones y duplicaciones pueden modificar grandes segmentos del cromosoma.
- Las inversiones y translocaciones dan lugar a una pequeña o ninguna pérdida de información genética.
- Los lugares frágiles son constricciones o brechas que aparecen en regiones particulares de los cromosomas con una predisposición a romperse en determinadas condiciones.
- El estudio de las series normales y anormales de cromosomas se conoce como citogenética.
Aneuploidía
La alteración en el número de cromosomas es denominada aneuploidía. La aneuploidía se define como la pérdida o ganancia de cromosomas completos en un individuo. Este fenómeno puede ocurrir en cualquiera de los cromosomas autosómicos (del 1 al 22) o sexuales (X e Y).
La ganancia de un cromosoma completo en una célula es denominada trisomía(2n+1), y en ese caso el cariotipo del individuo estaría formado por 47 cromosomas. Probablemente la trisomía más conocida sea el Síndrome de Down (trisomía del cromosoma 21). La pérdida de un cromosoma es denominada monosomía(2n-1) y el número de cromosomas de cada célula sería 45. La única monosomía viable en el hombre es la del cromosoma X, que origina en los individuos que la padecen el Síndrome de Turner.
En las células somáticas hay un mecanismo que inactiva a todos los cromosomas X menos uno, la ganancia o perdida de un cromosoma sexual en genoma diploide altera el fenotipo normal, dando lugar a los síndromes de Klinefelter o de Turner, respectivamente.
Tal variación cromosómica se origina como un error aleatorio durante la producción de gametos. La no disyunción es el fallo de los cromosomas o de las cromatidas en separarse y desplazarse a los polos opuestos en la meiosis. Cuando esto ocurre se desbarata la distribución normal de los cromosomas en los gametos. El cromosoma afectado puede dar lugar a gametos anormales con dos miembros o con ninguno. La fecundación de estos con un gameto haploide normal da lugar a zigotos con tres miembros (trisomía) o con solo uno (monosomía) de este cromosoma. La no disyunción da lugar a una serie de situaciones aneuploides autosómicas en la especie humana y en otros organismos.
Síndrome de Klinefelter
El síndrome de Klinefelter se considera la anomalía gonosómica más común en los humanos. Los afectados presentan un cromosoma “X” supernumerario lo que conduce a fallo testicular primario con infertilidad e hipoandrogenismo. A pesar de la relativa frecuencia del padecimiento en recién nacidos vivos, se estima que la mitad de los productos 47, XXY se abortan de manera espontánea.
Síndrome de Turner
El síndrome de Turner o Monosomía X es una enfermedad genética caracterizada por presencia de un solo 'cromosoma X'. La falta de cromosoma Y determina el sexo femenino de todos los individuos afectados, y la ausencia de todo o parte del segundo cromosoma X determina la falta de desarrollo de los caracteres sexuales primarios y secundarios. Esto confiere a las mujeres que padecen el síndrome de Turner un aspecto infantil e infertilidad de por vida.
Variaciones en estructura y ordenación de los cromosomas
El otro tipo de aberración cromosómica incluye cambios estructurales que eliminan, añaden o reordenan partes sustanciales de uno o más cromosomas, se encuentran las deleciones y las duplicaciones de genes o de parte de un cromosoma y las reordenaciones del material genético mediante las que segmentos de un cromosoma se invierten, se intercambian con un segmento de un cromosoma no homólogo o simplemente se transfieren a otro cromosoma. Los intercambios y las transferencias se denominan translocaciones, en las que la localización de un gen esta cambiada dentro del genoma.
Estos cambios estructurales se deben a una o más roturas distribuidas a lo largo del cromosoma, seguidas por la pérdida o la reordenación del material genético. Los cromosomas pueden romperse espontáneamente, pero la tasa de roturas puede aumentar en células expuestas a sustancias químicas o a radiación. Aunque los extremos normales de los cromosomas, los telómeros, no se fusionan fácilmente con extremos nuevos de cromosomas rotos o con otros telómeros, los extremos producidos en los puntos de rotura son cohesivos (“pegajosos”) y pueden reunirse con otros extremos rotos. Si la rotura y reunión no restablece las relaciones originales y si la alteración se produce en el plasma germinal, los gametos tendrán una reordenación estructural que será heredable.
Si la aberración se encuentra en un homólogo, pero no en el otro, se dice que los individuos son heterocigotos para la aberración. En tales casos se producen configuraciones raras en el apareamiento durante la sinapsis meiótica.
Si no hay pérdida o ganancia de material genético, los individuos que llevan la aberración en heterocigosis en uno de los dos homólogos probablemente no quedaran afectados en su fenotipo. Los complicados apareamientos de las ordenaciones dan lugar a menudo a gametos con duplicaciones o deficiencias de algunas regiones cromosómicas. Cuando esto ocurre, los descendientes de “portadores” de ciertas aberraciones tienen a menudo una mayor probabilidad de presentar cambios fenotípicos.
Translocaciones
Las translocaciones ocurren cuando un fragmento de ADN es transferido desde un cromosoma a otro no homólogo. Se incluyen:
- Traslocaciones recíprocas: Es una translocación balanceada. No hay pérdida o ganancia neta de material genético. Los individuos portadores de translocaciones recíprocas no suelen presentar ningún fenotipo. Sin embargo, estos individuos portadores tienen riesgo de producir descendencia con translocaciones desbalanceadas, que sí pueden estar asociadas a patologías o conducir al aborto del feto. También pueden ser un problema las translocaciones recíprocas de novo, si la rotura del cromosoma tiene lugar en genes importantes.
- Translocaciones desbalanceadas: Hay pérdida o ganancia de material genético respecto al genotipo silvestre. Pueden suponer un problema para el individuo portador de las mismas.
- Translocacion Robertsoniana: Son translocaciones "casi" equilibradas. Este tipo de mutación cromosómica tiene lugar con la fusión de los brazos largos de doscromosomas acrocéntricos. Los brazos cortos de ambos cromosomas se pierden. El cariotipo de los individuos con este tipo de translocaciones muestra 45 cromosomas, sin embargo no produce anomalía fenotípicas puesto que casi todo el material genético está presente. Las translocaciones Robertsonianas en un individuo pueden ser responsables de translocaciones desbalanceadas en su descendencia.
Mutaciones cromosómicas y cáncer
La mayoría de los tumores contienen varios tipos de mutaciones cromosómicas. Algunos tumores se asocian con deleciones, inversiones o translocaciones específicos.
- Las deleciones pueden eliminar o inactivar los genes que controlan el ciclo celular;
- Las inversiones y las translocaciones pueden causar rupturas en los genes supresores de tumores, fusionar genes que producen proteínas cancerígenas o mover genes a nuevas ubicaciones, donde quedan bajo la influencia de diferentes secuencias reguladoras.
- El papel de las mutaciones en el cáncer.
Las mutaciones en los genes regulatorios claves (los supresores de tumor y los protooncogenes) alteran el estado de las células y pueden causar el crecimiento irregular visto en el cáncer. Para casi todos los tipos de cáncer que se han estudiado hasta la fecha, parece que la transición de una célula sana y normal a una célula cancerosa es una progresión por pasos que requiere cambios genéticos en varios oncogenes y supresores de tumor diferentes. Esta es la razón por la cual el cáncer es mucho más prevalente en individuos de edades mayores. Para generar una célula cancerosa, una series de mutaciones deben ocurrir en la misma célula. Ya que la probabilidad de que cualquier gen sea mutado es muy baja, es razonable decir que la probabilidad de varias mutaciones en la misma célula es aún más improbable.
Mutaciones genómicas o numéricas

Son las mutaciones que afectan al número de cromosomas o todo el complemento cromosómico (todo el genoma).
- Poliploidía: Es la mutación que consiste en el aumento del número normal de “juegos de cromosomas” . Los seres poliploides pueden ser autopoliploides, si todos los juegos proceden de la misma especie, o alopoliploides, si proceden de la hibridación, es decir, del cruce de dos especies diferentes.
- Haploidía: Son las mutaciones que provocan una disminución en el número de juegos de cromosomas.
- Aneuploidía: Son las mutaciones que afectan solo a un número de ejemplares de un cromosoma o más, pero sin llegar a afectar al juego completo. Las aneuploidías pueden ser monosomías, trisomías, tetrasomías, etc, cuando en lugar de dos ejemplares de cada tipo de cromosomas, que es lo normal, hay o solo uno, o tres, o cuatro, etc. Entre las aneuplodías podemos encontrar diferentes tipos de trastornos genéticos en humanos como pueden ser:
- Trisomía 21 o Síndrome de Down que tienen 47 cromosomas.
- Trisomía 18 o Síndrome de Edwards. También tienen 47 cromosomas.
- Trisomía 13 o Síndrome de Patau.
- Monosomía X o Síndrome de Turner.
- Trisomía sexual XXX o Síndrome del triple X.
- Trisomía sexual XXY o Síndrome de Klinefelter.
- Trisomía sexual XYY o Síndrome del doble Y.
- Cromosoma extra Síndrome de Down.
Mutaciones génicas o moleculares
Son las mutaciones que alteran la secuencia de nucleótidos del ADN. Estas mutaciones pueden llevar a la sustitución de aminoácidos en las proteínas resultantes (se denominan mutaciones no sinónimas). Un cambio en un solo aminoácido puede no ser importante si es conservativo y ocurre fuera del sitio activo de la proteína. Así, existen las denominadas mutaciones sinónimas o "mutaciones silenciosas" en las que la mutación altera la base situada en la tercera posición del codón pero no causa sustitución aminoacídica debido a la redundancia del código genético. El aminoácido insertado será el mismo que antes de la mutación. También, en el caso de lasmutaciones neutras, el aminoácido insertado es distinto pero con unas propiedades fisicoquímicas similares, por ejemplo la sustitución de glutámico por aspártico puede no tener efectos funcionales en la proteína debido a que los dos son ácidos y similares en tamaño. También podrían considerarse neutras aquellas mutaciones que afecten a zonas del genoma sin función aparente, como las repeticiones en tándem o dispersas, las zonas intergénicas y los intrones.8
De lo contrario, la mutación génica o también llamada puntual, puede tener consecuencias severas, como por ejemplo:
- La sustitución de valina por ácido glutámico en la posición 6 de la cadena polipeptídica de la beta-globina da lugar a la enfermedad anemia falciforme en individuos homocigóticos debido a que la cadena modificada tiene tendencia a cristalizar a bajas concentraciones de oxígeno.
- Las proteínas del colágeno constituyen una familia de moléculas estructuralmente relacionadas que son vitales para la integridad de muchos tejidos incluidos la piel y los huesos. La molécula madura del colágeno está compuesta por 3 cadenas
polipeptídicas unidas en una triple hélice. Las cadenas se asocian primero por su extrempo C-terminal y luego se enroscan hacia el extremo N-terminal. Para lograr este plegado, las cadenas de colágeno tienen una estructura repetitiva de 3 aminoácidos: glicina - X - Y (X es generalmente prolina y Y puede ser cualquiera de un gran rango de aminoácidos). Una mutación puntual que cambie un solo aminoácido puede distorsionar la asociación de las cadenas por su extremo C-terminal evitando la formación de la triple hélice, lo que puede tener consecuencias severas. Una cadena mutante puede evitar la formación de la triple hélice, aun cuando haya 2 monómeros de tipo salvaje. Al no tratarse de una enzima, la pequeña cantidad de colágeno funcional producido no puede ser regulada. La consecuencia puede ser la condición dominante letal osteogénesis imperfecta.
Bases moleculares de la mutación génica
- Mutación por sustitución de bases: Se producen al cambiar en una posición un par de bases por otro (son las bases nitrogenadas las que distinguen los nucleótidos de una cadena). Distinguimos dos tipos que se producen por diferentes mecanismos bioquímicos:8
- Mutaciones transicionales o simplemente transiciones, cuando un par de bases es sustituido por su alternativa del mismo tipo. Las dos bases púricas son adenina (A) y guanina (G), y las dos pirimídicas son citosina (C) y timina (T). La sustitución de un par AT, por ejemplo, por un par GC, sería una transición.
- Mutaciones transversionales o transversiones, cuando un par de bases es sustituida por otra del otro tipo. Por ejemplo, la sustitución del par AT por TA o por CG.
- Mutaciones de corrimiento estructural, cuando se añaden o se quitan pares de nucleótidos alterándose la longitud de la cadena. Si se añaden o quitan pares en un número que no sea múltiplo de tres (es decir si no se trata de un número exacto de codones), las consecuencias son especialmente graves, porque a partir de ese punto, y no solo en él, toda la información queda alterada. Hay dos casos:
- Mutación por pérdida o deleción de nucleótidos: en la secuencia de nucleótidos se pierde uno y la cadena se acorta en una unidad.
- Mutación por inserción de nuevos nucleótidos: Dentro de la secuencia del ADN se introducen nucleótidos adicionales, interpuestos entre los que ya había, alargándose correspondientemente la cadena.8
- Mutaciones en los sitios de corte y empalme, montaje o ayuste (Splicing)
Las mutaciones de corrimiento del marco de lectura también pueden surgir por mutaciones que interfieren con el ayuste del ARN mensajero. El comienzo y final de cada intrón en un gen están definidos por secuencias conservadas de ADN. Si un nucleótido muta en una de las posiciones altamente conservada, el sitio no funcionará más, con las consecuencias predecibles para el ARNm maduro y la proteína codificada. Hay muchos ejemplos de estas mutaciones, por ejemplo, algunas mutaciones en el gen de la beta globina en la beta talasemia son causadas por mutaciones de los sitios de ayuste.
Mutaciones espontáneas o inducidas
Las mutaciones pueden ser espontáneas o inducidas. Las primeras son aquellas que surgen normalmente como consecuencia de errores durante el proceso de replicación del ADN. Tales errores ocurren con una probabilidad de 10−7 en células haploides y 10−14 en diploides.8
Mutaciones inducidas
Las mutaciones inducidas surgen como consecuencia de la exposición a mutágenos químicos o biológicos o a radiaciones. Entre los mutágenos químicos se pueden citar:
- los análogos de bases del ADN (como la 2-aminopurina), moléculas que se parecen estructuralmente a las bases púricas o pirimidínicas pero que muestran propiedades de apareamiento erróneas;
- los agentes alquilantes como la nitrosoguanidina, que reacciona directamente con el ADN originando cambios químicos en una u otra base y produciendo también apareamientos erróneos;
- y, por último, los agentes intercalantes como las acridinas, que se intercalan entre 2 pares de bases del ADN, separándolas entre sí.
Como mutágenos biológicos podemos considerar la existencia de transposones o virus capaces de integrarse en el genoma.
Las radiaciones ionizantes (rayos X, rayos cósmicos y rayos gamma) y no ionizantes (sobre todo la radiación ultravioleta) también inducen mutaciones en el ADN; las primeras se originan por los radicales libres que reaccionan con el ADN inactivándolo, y las segundas aparecen como consecuencia de la formación de dímeros de pirimidina en el ADN, es decir, como consecuencia de la unión covalente de 2 bases pirimidínicas adyacentes.
Un agente utilizado a menudo para inducir mutaciones (mutagénesis) en organismos experimentales es el EMS (sulfato de etilmetano). Este mutágeno puede alterar la secuencia del DNA de diversas maneras como modificar químicamente las bases de G en DNA. Esta alteración en la secuencia de un gen se conoce como mutación puntual.
Mutaciones espontáneas
Las principales causas de las mutaciones que se producen de forma natural o normal en las poblaciones son tres: los errores durante la replicación del ADN, las lesiones o daños fortuitos en el ADN y la movilización en el genoma de los elementos genéticos transponibles.
Errores en la replicación
Durante la replicación del ADN pueden ocurrir diversos tipos de errores que conducen a la generación de mutaciones. Los tres tipos de errores más frecuentes son:
- La tautomería: las bases nitrogenadas se encuentran habitualmente en su forma cetónica y con menos frecuencia aparecen en su forma tautomérica enólica o imino. Las formas tautoméricas o enólicas de las bases nitrogenadas (A*, T*, G* y C*) muestran relaciones de apareamiento distintas que las formas cetónicas: A*-C, T*-G, G*-T y C*-A. El cambio de la forma normal cetónica a la forma enólica produce transiciones. Los errores en el apareamiento incorrecto de las bases nitrogenadas pueden ser detectados por la función correctora de pruebas de la ADN polimerasa III.
- Las mutaciones de cambio de fase o pauta de lectura: se trata de inserciones o deleciones de uno o muy pocos nucleótidos. Según un modelo propuesto por Streisinger, estas mutaciones se producen con frecuencia en regiones con secuencias repetidas. En las regiones con secuencias repetidas, por ejemplo, TTTTTTTTTT..., o por ejemplo, GCGCGCGCGCGCG...., durante la replicación se puede producir el deslizamiento de una de las dos hélices (la hélice molde o la de nueva síntesis) dando lugar a lo que se llama "apareamiento erróneo deslizado". El deslizamiento de la hélice de nueva síntesis da lugar a una adición, mientras que el deslizamiento de la hélice molde origina una deleción. En el gen lac I (gen estructural de la proteína represora) de E. coli se han encontrado puntos calientes (regiones en las que la mutación es muy frecuente) que coinciden con secuencias repetidas: un ejemplo es el punto caliente CTGG CTGG CTGG.
- Deleciones y duplicaciones grandes: las deleciones y duplicaciones de regiones relativamente grandes también se han detectado con bastante frecuencia en regiones con secuencias repetidas. En el gen lac I de E. coli se han detectado deleciones grandes que tienen lugar entre secuencias repetidas. Se cree que estas mutaciones podrían producirse por un sistema semejante al propuesto por Streisinger ("Apareamiento erróneo deslizado") o bien por entrecruzamiento desigual.8
Lesiones o daños fortuitos en el ADN



Pueden darse tres tipos de daños fortuitos en el ADN:
- La despurinización consiste en la ruptura del enlace glucosídico entre la base nitrogenada y el azúcar al que está unida con pérdida de una adenina o de una guanina . Como consecuencia aparecen sitios apurínicas (o sea, sin bases púricas). Existe un sistema de reparación de este tipo de lesiones en el ADN. Este tipo de lesión es la más recurrente o frecuente: se estima que se produce una pérdida de 10.000 cada 20 horas a 37 °C.
- La desaminación consiste en la pérdida de grupos amino. La citosina por desaminación se convierte en uracilo y el uracilo empareja con adenina produciéndose transiciones: GC→AT. El uracilo no forma parte del ADN, existiéndo un enzima llamada glucosidasa de uracilo encargada de detectar la presencia de este tipo de base en el ADN y retirarlo. Al retirar el uracilo se produce una sede o sitio apirimidínica. La 5-Metil-Citosina (5-Me-C) por desaminación se convierte en Timina (T). La Timina (T) es una base normal en el ADN y no se retira, por tanto estos errores no se reparan. Este tipo de mutación también genera transiciones.
- Los daños oxidativos en el ADN. El metabolismo aeróbico produce radicales superoxido O2, peróxido de hidrógeno H2O2 e hidroxilo. Estos radicales producen daños en el ADN, y una de las principales alteraciones que originan es la transformación de la guanina en 8-oxo-7,8-dihidro-desoxiguanina que aparea con la Adenina. La 8-oxo-7,8-dihidro-desoxiguanina recibe el nombre abreviado de 8-oxo-G. Esta alteración del ADN produce transversiones: GC→TA.8
Elementos genéticos transponibles
Los elementos genéticos transponibles son secuencias de ADN que tienen la propiedad de cambiar de posición dentro del genoma, por tal causa también reciben el nombre de elementos genéticos móviles. Por tanto, cuando cambian de posición y abandonan el lugar en el que estaban, en ese sitio, se produce un deleción o pérdida de bases. Si el elemento transponible estaba insertado en el interior de un gen, puede que se recupere la función de dicho gen. De igual forma, si el elemento genético móvil al cambiar de posición se inserta dentro de un gen se produce una adición de una gran cantidad de nucleótidos que tendrá como consecuencia la pérdida de la función de dicho gen. Por consiguiente, los elementos genéticos transponibles producen mutaciones.
Su existencia fue propuesta por Barbara McClintock (1951 a 1957) en el maíz. Sin embargo, su existencia no se demostró hasta mucho más tarde en bacterias. En el fenómeno de la transposición no se ha encontrado una relación clara entre la secuencia de la sede donadora (lugar en el que está el transposón) y la sede aceptora (lugar al que se incorpora el transposón). Algunos transposones muestran una preferencia por una determinada región (zona de 2000 a 3000 pares de bases), pero dentro de ella parecen insertarse al azar.
- Transposones en Bacterias
En Bacterias existen dos tipos de transposones:
- Transposón Simple, Secuencia de Inserción o Elemento de Inserción (IS): los transposones simples contienen una secuencia central con información para la transposasa y en los extremos una secuencia repetida en orden inverso. Esta secuencia repetida en orden inverso no es necesariamente idéntica, aunque muy parecida. Cuando un transposón simple se integra en luna determinado punto del ADN aparece una repetición directa de la secuencia diana (5-12 pb).
- Transposón Compuesto (Tn): contienen un elemento de inserción (IS) en cada extremo en orden directo o inverso y una región central que además suele contener informaciión de otro tipo. Por ejemplo, los Factores de transferencia de resistencia (RTF), poseen información en la zona central para resistencia a antibióticos (cloranfenicol, kanamicina,tetraciclina, etc.).
Tanto los elementos IS como los transposones compuestos (Tn) tienen que estar integrados en otra molécula de ADN, el cromosoma principal bacteriano o en un plasmidio, nunca se encuentran libres.
- Transposones en eucariotas
- Transposones en plantas
Los transposones fueron descubiertos por Barbara McClintock (entre 1951 y 1957) en maíz, sin embargo, cuando postuló su existencia la comunidad científica no comprendió adecuadamente sus trabajos. Años más tarde, ella misma comparó los "elementos controladores" que había descrito (elementos cromosómicos transponibles) de maíz con los transposones de los plasmidios. Sus trabajos recibieron el Premio Nobel en 1983.
Dentro de las familias de elementos controladores de maíz se pueden distinguir dos clases:
-
- Los elementos autónomos: capaces de escindirse de la sede donadora y transponerse.
-
- Los elementos no autónomos: son estables, y solamente se vuelven inestables en presencia de los autónomos en posición trans.
En el sistema Ac-Ds (Activador-Disociación) estudiado por McClintock, Ac es el elemento autónomo y Ds es el elemento no autónomo. Además del sistema Ac-Ds en maíz se han descrito otros sistemas como el Mu (Mutador), sistema Spm(Supresor-Mutador), sistema R-stippled y sistema MrRm. También se han encontrado transposones en otras especies de plantas, tales como en la "boca de dragón" o "conejito" (Anthirrhinum majus), en Petunia y en soja (Glycine max), etc..
- Transposones en mamíferos
En mamíferos se conocen tres clases de secuencias que son capaces de transponerse o cambiar de posición a través de un ARN intermediario:
-
- Retrovirus endógenos: semejantes a los retrovirus, no pueden infectar nuevas células y están restringidos a un genoma, pero pueden transponerse dentro de la célula. Poseen largas secuencias repetidas en los extremos (LTR), genes env (con información para la proteína de la cubierta) y genes que codifican para la trasnrciptasa inversa, como los presentes en retrovirus.
-
- Retrotransposones o retroposones: carecen de LTR y de los genes env (con información para la proteína de la cubierta) de retrovirus. Contienen genes para la transcriptasa inversa y pueden transponerse. Tienen una secuencia rica en pares A-T en un extremo. Un ejemplo, son los elementos LINE-1 (elementos largos dispersos) en humanos y ratones.
-
- Retropseudogenes: carecen de genes para la transcriptasa inversa y por consiguiente son incapaces de transponerse de forma independiente, aunque si pueden cambiar de posición en presencia de otros elementos móviles que posean información para la trasncriptasa inversa. Poseen una región rica en pares A-T en un extremo y los hay de dos tipos:
- Pseudogenes procesados: están en bajo número de copias y derivan de genes transcritos por la ARN Poilimerasa II, siendo genes que codifican para polipéptidos. Estos pseudogenes procesados carecen de intrones.
- SINES (elementos cortos dispersos): están en alto número de copias en mamíferos. Dos ejemplos son la secuencia Alu de humanos y B1 de ratón, que derivan de genes transcritos por la ARN polimerasa III utilizando un promotor interno.
- Retropseudogenes: carecen de genes para la transcriptasa inversa y por consiguiente son incapaces de transponerse de forma independiente, aunque si pueden cambiar de posición en presencia de otros elementos móviles que posean información para la trasncriptasa inversa. Poseen una región rica en pares A-T en un extremo y los hay de dos tipos:
La secuencia Alu es la más abundante en el genoma humano, existiendo 750.000 copias dispersas por el genoma, aproximadamente existe una copia cada 4000 pb. Esta secuencia posee un contenido relativamente alto en (G+C) y presenta una elevada homología (70-80 %) con la secuencia B1 de ratón. Se la denomina secuencia Alu por poseer en su interior una diana para la endonucleasa de restricción Alu. Las secuencias Alu humanas tienen alrededor de 280 pb y están flanqueadas por repeticiones directas cortas (6-18 pb). Una secuencia típica Alu es un dímero repetido en tandem, la unidad que se repite tiene un tamaño aproximado de 120 pb y va seguida de una corta secuencia rica en pares A-T. Sin embargo, existe una asimetría en las unidades repetidas, de manera que la segunda unidad contiene una secuencia de 32 pb ausente en la primera. Las unidades repetidas de la secuencia Alu muestran un elevado parecido con la secuencia del ARN 7SL, un componente que juega un papel importante en el transporte de las proteínas a través de la membrana del retículo endoplasmático.
Dominancia y recesividad de las mutaciones
La mayoría de las mutaciones son recesivas
La mayoría de las mutaciones son recesivas debido a que la mayor parte de los genes codifica para enzimas. Si un gen esta inactivo se produce una reducción en el nivel de actividad de la enzima que puede no ser superior al 50 % ya que el nivel de transcripción de los genes residuales puede aumentarse por regulación en respuesta a cualquier aumento en la concentración del sustrato. Asimismo, la proteína en si misma puede estar sujeta a regulación (por fosforilación, por ejemplo) de tal forma que su actividad pueda ser aumentada para compensar cualquier falta en el número de moléculas. En cualquier caso, a menos que la enzima controle la velocidad del paso limitante en la ruta bioquímica, una reducción en la cantidad de producto puede no importar. l fenotipo. Esta enfermedad es causada por mutaciones en el gen que codifica para la enzima fenilalanina hidroxilasa, la cual convierte el aminoácido fenilalanina a tirosina. Si un individuo es homocigota para alelos que eliminen completamente cualquier actividad de esta enzima, la fenilalanina no podrá ser metabolizada y aumentará sus niveles en sangre hasta un punto en el cual comienza a ser dañina para el cerebro en desarrollo. Es de rutina determinar esta condición en los recién nacidos mediante el análisis de una pequeña gota de sangre (Test Guthrie). Este estudio ha revelado que existen pocas personas con una condición conocida como Hiperfenilalaninemia Benigna. Estos individuos tienen niveles moderadamente altos fenilalanina en sangre. Sus niveles de fenilalanina hidroxilasa constituyen aproximadamente el 5 % del normal. A pesar de esto, son aparentemente perfectamente saludables y no sufren de las anormailidades cerebrales causadas por la falta total de la actividad enzimática.
Mutaciones dominantes
Haploinsuficiencia
En este caso, la cantidad de producto de un gen no es suficiente para que el metabolismo sea el normal. Quizás la enzima producida sea la responsable de regular la velocidad del paso limitante en una reacción de una ruta metabólica. La telangiectasia hemorrágica hereditaria es una displasia vascular autosómica dominante que lleva a telangiectasias y malformaciones arteriovenosas de la piel, mucosas y vísceras, provocando ocasionalmente la muerte por sangrados incontrolados. Está causada por una mutación en el gen ENG, que codifica para la endoglina, proteína receptora del factor beta transformante de crecimiento (TGF-beta). Quizás el TGF-beta no sea capaz de ejercer un efecto suficiente en las células cuando solo está presente la mitad de la cantidad normal del receptor.9 10 11
Efecto dominante negativo
Ciertas enzimas tiene una estructura multimérica (compuesta por varias unidades) y la inserción de un componente defectuoso dentro de esa estructura puede destruir la actividad de todo el complejo. El producto de un gen defectuoso, entonces, interfiere con la acción del alelo normal. Ejemplos de este efecto son las mutaciones que causan la osteogénesis imperfecta y ciertos tumores intestinales.12 13
Ganancia de función
Es imposible imaginar que por una mutación un gen pueda ganar una nueva actividad, pero quizá el sitio activo de una enzima pueda ser alterado de tal forma que desarrolle especificidad por un nuevo sustrato. Si esto es así, cómo puede ocurrir la evolución? Ejemplos en genética humana de genes con 2 alelos tan diferentes son raras pero un ejemplo está dado por el locus ABO. La diferencia entre los loci A y B está determinada por 7 cambios nucleotídicos que llevaron a cambios en 4 aminoácidos. Probablemente solo uno de estos cambios es responsable del cambio en especificidad entre las enzimas alfa-3-N-acetil-D-galactosaminiltransferasa (A) y alfa-3-D-galactosiltransferasa. También hay muchos ejemplos de la evolución humana donde muchos genes se han duplicado y en consecuencia han divergido en sus especificidades por el sustrato. En el cromosoma 14 hay un pequeño grupo de 3 genes relacionados, alfa-1-antitripsina (AAT), alfa-1-antiquimotripsina (ACT) y un gen relacionado que ha divergido de tal forma que probablemente ya no sea funcional. Las relaciones estructurales entre AAT y ACT son muy obvias y ambos son inhibidores de proteasas, pero ahora claramente cumplen roles levemente diferentes debido a que tienen diferentes actividades contra un rango de proteasas y están bajo una regulación diferente.
Dominancia a nivel organísmico pero recesividad a nivel celular
Algunos de los mejores ejemplos de esto se encuentran en el área de la genética del cáncer. Un ejemplo típico sería el de un gen supresor de tumor como enretinoblastoma.
Tasas de mutación
Las tasas de mutación han sido medidas en una gran variedad de organismos. En mamíferos la tasa de mutación de 1 en  bases núcleotídicas,14 mientras que, en el otro extremo de la escala los virus de ARN tienen una tasa de mutación del orden de 1 en
bases núcleotídicas,14 mientras que, en el otro extremo de la escala los virus de ARN tienen una tasa de mutación del orden de 1 en 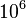 .15 La cantidad de mutaciones tiene relación con el tipo de enzima involucrada en la copia del material genético. Esta enzima (ADN o ARN Polimerasa, según el caso) tiene distintas tasas de error y esto incide directamente en el número final de mutaciones. A pesar de que la incidencia de las mutaciones es relativamente grande en relación con el número de organismos de cada especie, la evolución no depende solo de las mutaciones que surgen en cada generación, sino de la interacción de toda esta acumulación de variabilidad con la selección natural y la deriva genética durante la evolución de las especies.
.15 La cantidad de mutaciones tiene relación con el tipo de enzima involucrada en la copia del material genético. Esta enzima (ADN o ARN Polimerasa, según el caso) tiene distintas tasas de error y esto incide directamente en el número final de mutaciones. A pesar de que la incidencia de las mutaciones es relativamente grande en relación con el número de organismos de cada especie, la evolución no depende solo de las mutaciones que surgen en cada generación, sino de la interacción de toda esta acumulación de variabilidad con la selección natural y la deriva genética durante la evolución de las especies.
bases núcleotídicas,14 mientras que, en el otro extremo de la escala los virus de ARN tienen una tasa de mutación del orden de 1 en .15 La cantidad de mutaciones tiene relación con el tipo de enzima involucrada en la copia del material genético. Esta enzima (ADN o ARN Polimerasa, según el caso) tiene distintas tasas de error y esto incide directamente en el número final de mutaciones. A pesar de que la incidencia de las mutaciones es relativamente grande en relación con el número de organismos de cada especie, la evolución no depende solo de las mutaciones que surgen en cada generación, sino de la interacción de toda esta acumulación de variabilidad con la selección natural y la deriva genética durante la evolución de las especies.Mutaciones y polimorfismos
Las mutaciones pueden considerarse patológicas o anormales, mientras que los polimorfismos son variaciones normales en la secuencia del ADN entre unos individuos a otros y que superan el uno por ciento en la población, por lo que no puede considerarse patológico. La mayoría de los polimorfismos proceden de mutaciones silentes.
Contribución de las mutaciones al organismo
La contribución de las mutaciones a los tejidos es diferente, lo que puede deberse a las distintas tasas de mutación por división celular y al distinto número de divisiones celulares en cada tejido.
Además, sabiendo los procesos mutacionales, las tasas de mutación y el proceso de desarrollo de los tejidos, se puede conocer la historia de las células individuales. Para lo que hay que basarse en la secuenciación del genoma celular.
Mutación y evolución
Las mutaciones son la materia prima de la evolución biológica. La evolución tiene lugar cuando una nueva versión de un gen, que originalmente surge por una mutación, aumenta su frecuencia y se extiende a la especie gracias a la selección natural o a tendencias genéticas aleatorias (fluctuaciones casuales en la frecuencia de los genes). Antes se pensaba que las mutaciones dirigían la evolución, pero en la actualidad se cree que la principal fuerza directora de la evolución es la selección natural, no las mutaciones. No obstante, sin mutaciones las especies no evolucionarían.
La selección natural actúa para incrementar la frecuencia de las mutaciones ventajosas, que es como se produce el cambio evolutivo, ya que esos organismos con mutaciones ventajosas tienen más posibilidades de sobrevivir, reproducirse y transmitir las mutaciones a su descendencia.
La selección natural actúa para eliminar las mutaciones desventajosas; por tanto, está actuando continuamente para proteger a la especie de la decadencia mutacional. Sin embargo, la mutación desventajosa surge a la misma velocidad a la que la selección natural la elimina, por lo que las poblaciones nunca están completamente limpias de formas mutantes desventajosas de los genes. Esas mutaciones que no resultan ventajosas pueden ser el origen de enfermedades genéticas que pueden transmitirse a la siguiente generación.
La selección natural no actúa sobre las mutaciones neutrales, pero las mutaciones neutrales pueden cambiar de frecuencia por procesos aleatorios. Existen controversias sobre el porcentaje de mutaciones que son neutrales, pero generalmente se acepta que, dentro de las mutaciones no neutras, las mutaciones desventajosas son mucho más frecuentes que las mutaciones ventajosas. Por tanto, la selección natural suele actuar para reducir el porcentaje de mutaciones al mínimo posible; de hecho, el porcentaje de mutaciones observado es bastante bajo.
Mutación y cáncer
El cáncer está causado por alteraciones en oncogenes, genes supresores de tumores y/o genes de micro ARN. Un solo cambio genético es usualmente insuficiente para que se desarrolle un tumor maligno. La mayor parte de la evidencia indica que tal desarrollo involucra un proceso de varios pasos secuenciales en los cuales ocurren alteraciones en varios, frecuentemente muchos, de estos genes.16 Un oncogén es un gen que, cuando es desregulado, participa en el inicio y desarrollo del cáncer. Las mutaciones génicas que dan como resultado la activación de los oncogenes incrementan la posibilidad de que una célula normal se convierta en una célula tumoral. Desde la década de los '70 se han identificado docenas de oncogenes en los seres humanos. Los oncogenes, al menos en sentido figurado, son los perpetuos antagonistas de los genes supresores tumorales, los cuales actúan previniendo el daño del ADN y mantienen las funciones celulares bajo un equilibrado control. Existe mucha evidencia que apoya la noción de que la pérdida o inactivación por mutaciones puntuales de los genes supresores de tumores puede llevar a una célula a transformarse en cancerosa.17 Los oncogenes se originan a partir de mutaciones en genes normales, llamados proto-oncogenes. Los proto-oncogenes usualmente codifican para proteínas que ayudan a regular el ciclo celular o la diferenciación celular y se hallan frecuentemente involucrados en la transducción de señal y en la ejecución de señales mitogénicas.<.18 Se ha descubierto, por otro lado, que los micro ARNs (pequeños ARNs de 20 a 25 nucléotidos de longitud) pueden controlar la expresión de los oncogenes regulándolos negativamente.19 Por esa razón, las mutaciones en los micro ARNs pueden llevar a la activación de los oncogenes.20
Hipermutación somática
La hipermutación somática (o SHM, por sus siglas en inglés) es un mecanismo celular, que forma parte del modo en cómo se adapta el sistema inmune a nuevos elementos extraños (por ejemplo bacterias). Su función es diversificar los receptores que usa el sistema inmunitario para reconocer elementos extraños (antígeno) y permite al sistema inmune adaptar su respuesta a las nuevas amenazas que se producen a lo largo de la vida de un organismo.21 La hipermutación somática implica un proceso de mutación programada que afecta a las regiones variables de los genes de inmunoglobulina. A diferencia de muchos otros tipos de mutación, la SHM afecta solo a células inmunitarias individuales y sus mutaciones, por lo tanto, no se trasmiten a la descendencia.21
Diferentes tipos de mutación
La mutación se define tradicionalmente como una modificación en la información genética, producida por un cambio brusco y de tipo hereditario, interviniendo uno o varios caractéres.
Sin embargo, la puesta en evidencia del ADN como soporte químico de la información genética y la posibilidad de acceder al conocimiento específico de la secuencia de nucleótidos que caracteriza cada cromosoma ha llevado a proponer una nueva definición: Todo cambio que afecta la secuencia de nucleótidos es una mutación.22
Mutaciones y genética de poblaciones
Además, a nivel de la genética de poblaciones se define como un error en la reproducción conforme al mensaje hereditario. Ella va a transformar un alelo en otro, nuevo o ya existente en la población. El rol de la mutación en la evolución es primordial, porque es la única fuente de genes nuevos. Sin embargo, una vez que un nuevo gen ha aparecido en la población, ya no es él mismo quien va a determinar su futuro: si este nuevo alelo es más favorable o desfavorable que los antiguos, será la selección natural la que va a determinar la evolución posterior de su frecuencia en la población.23
A nivel de población, la persistencia depende de la mantención de la información genética. Para lograr esto, los organismos intentan disminuir la tasa de mutación y limitar las mutaciones deletéreas. Sin embargo, la adaptación a nuevas situaciones necesita un cierto nivel de variación genética para obtener mutaciones raras y benéficas. El número de mutaciones de una población es determinado por el tamaño de ella, además de la tasa de mutación del organismo. En consecuencia, para todo tamaño de población determinado, un organismo deberá desarrollar una tasa de mutación que optimice entre las mutaciones deletéreas comunes, y las raras mutaciones beneficiosas, que aumentan la adaptación a largo plazo. La relación óptima entre costo y beneficio deberá cambiar de acuerdo a las circunstancias y los hábitos de vida. Una tasa de mutación elevada podría ser más costosa para un organismo bien adaptado a su medioambiente constante, que para un organismo mal adaptado a un medioambiente que está en continuo cambio.24 De cualquier manera y en general, la tasa de mutación es minimizada por la selección. Hay, por otro lado, argumentos teóricos que muestran que las mutaciones pueden ser seleccionadas positivamente por el hecho de crecer en un medioambiente determinado, donde la selección necesita de mutantes raros repetidos y que la variabilidad genética es limitada. Esto sucede cuando la población es pequeña y los mutantes raros pueden ofrecer una ventaja selectiva (por ejemplo resistencia a los antibióticos) más importante que el costo selectivo para la adaptación.
Por ejemplo, en el caso de VIH, numerosas mutaciones aleatorias se producen a cada ciclo de la replicación viral, debido a la poca fidelidad que posee la transcriptasa inversa durante la transcripción. Algunas de estas mutaciones serán seleccionadas, por la presión que ejercen los Linfocitos T Citotóxicos (CTL) específicos para los epítopos salvajes. O las respuestas citotóxicas tempranas parecen tener una actividad anti-viral más eficaz, y el escape a esta respuesta explicaría la progresión viral.2526
Tipos de mutación en el VIH
Diferentes tipos de mutaciones pueden perturbar la presentación de moléculas del CMHI. Mutaciones a nivel de regiones colindantes de los epítopes van a intervenir con la capacidad de separación de proteínas virales por el proteosoma o con la capacidad de transporte celular. De la misma manera, mutaciones que suceden en los epítopes mismos, disminuyen la respuesta citotóxica específica para los CTL. Si estas mutaciones conciernen los residuos de anclaje, ellas podrían provocar una inhibición completa de la unión del péptido con las moléculas de CMHI.
En fin, las mutaciones relacionadas con los aminoácidos relacionados con los residuos de anclaje en los epítopes pueden igualmente modificar la interacción del péptido con la molécula del CMHI por motivos de conformación espacial. Si la unión CMHI-Péptido no es estable, el complejo es separado antes de la unión con el TCR (T Cell Receptor) y el reconocimiento del péptido por los linfocitos T citotóxicos no se llevará a cabo. Es así como el virus de VIH está obligado a estar en un permanente equilibrio entre las mutaciones de escape a la respuesta inmune y el costo funcional para él que podrían estar ligadas a estas mutaciones, como una dismución en la adaptación o de su poder infectante. Por otro lado, ha sido demostrado que en el caso de la respuesta por los CTL, las mutaciones ocurridas en regiones funcionales importantes conducirían a la no viabilidad de estos mutantes. Por ejemplo, mutaciones de escape a CTL en regiones codantes Gag p-24 van a producir una disminución significativa en la adaptación viral, por el contrario una mutación de escape en las regiones codantes Env gp-120 no tienen efecto en la adaptación viral.
No hay comentarios:
Publicar un comentario