Farmacocinética
El coeficiente de reparto (K) de una sustancia, también llamado coeficiente de distribución (D), o coeficiente de partición (P), es el cociente o razón entre lasconcentraciones de esa sustancia en las dos fases de la mezcla formada por dos disolventes inmiscibles en equilibrio. Por tanto, ese coeficiente mide la solubilidaddiferencial de una sustancia en esos dos disolventes.
![K = \frac{[sustancia]_1}{[sustancia]_2} \quad \quad](https://upload.wikimedia.org/math/4/7/9/4790c00ee59443abe59900a793a96cf1.png)
Donde [sustancia]1 es la concentración de la sustancia en el primer disolvente y, análogamente [sustancia]2 es la concentración de la misma sustancia en el otro disolvente.
Es un término muy usado en Bioquímica, Geoquímica, Química orgánica y en Química farmacéutica. En las dos últimas ciencias los disolventes más usados son el aguay un disolvente hidrófobo como el octanol (octan-1-ol, o n-octanol). En este caso recibe el nombre de coeficiente de reparto octanol-agua (Pow). El coeficiente de reparto indica el carácter hidrófilo o hidrófobo de una sustancia, es decir su mayor o menor tendencia a disolverse en disolventes polares (como el agua) o en disolventes apolares (como los disolventes orgánicos). Los coeficientes de partición o reparto son usados, por ejemplo, para estimar la distribución de fármacos en el cuerpo. Los fármacos con elevados coeficientes de partición son hidrófobos y se distribuyen preferentemente en entornos hidrófobos como las bicapas lipídicas de las células, mientras que los fármacos con coeficientes de reparto bajos son hidrófilos y se encuentran preferentemente en los entornos hidrófilos como el suero sanguíneo.
Definición legal
Según el Real Decreto 363/1995, de 10 de Marzo, por el que se aprueba el reglamento sobre notificación de sustancias nuevas y clasificación, envasado y etiquetado de sustancias peligrosas,1 en su Anexo V, parte A, apartado A-7:
1.2. DEFINICIONES Y UNIDADES
El coeficiente de reparto (P) se define como la relación de las concentraciones en equilibrio (ci) de una sustancia disuelta en un sistema bifásico consistente en dos disolventes considerablemente inmiscibles. En el caso del n-octanol y del agua:
-
-
- Pow = (C n-octanol / C agua )
-
El coeficiente de reparto (P) es, pues, el cociente de dos concentraciones: se indica generalmente en forma de su logaritmo decimal (log P).
Coeficiente de reparto y log P
El coeficiente de reparto de una sustancia es un cociente o proporción entre las concentraciones de un compuesto no-ionizado entre los dos disolventes. Para medir el coeficiente de reparto de solutos ionizables, el pH de la fase acuosa se ajusta de tal modo que la forma predominante del compuesto esté en la forma no-ionizada. Ellogaritmo del cociente entre las concentraciones del soluto no-ionizado en los disolventes se llama log P y se presenta en forma logarítmica porque el rango de valores que puede tomar es muy amplio:
![log\ P_{octanol/agua} = log\Bigg(\frac{\big[soluto\big]_{octanol}}{\big[soluto\big]_{agua}^{no-ionizado}}\Bigg)](https://upload.wikimedia.org/math/4/a/3/4a3e7274b9ea9b7b4e7ab65fc1266bee.png)
Se puede consultar un conjunto de datos de coeficientes de reparto octanol-agua, para ver su variabilidad.
Coeficiente de distribución y log D
El coeficiente de distribución es el cociente o razón de la suma de las concentraciones de todas las formas del compuesto (ionizadas y no-ionizadas) en cada una de las dos fases. Para medidas del coeficiente de distribución, el pH de la fase acuosa está tamponado para un valor específico tal que el pH no se disturbe de modo significativo por la introducción del compuesto. El logaritmo del cociente entre la suma de las concentraciones de todas las formas del compuesto (ionizadas y no-ionizadas) en un disolvente, y la suma de las concentraciones de todas sus formas en el otro disolvente se llama Log D y se presenta en forma logarítmica porque el rango de valores que puede tomar es muy amplio:
![log\ D_{octanol/agua} = log\Bigg(\frac{\big[soluto\big]_{octanol}}{\big[soluto\big]_{agua}^{ionizado}+\big[soluto\big]_{agua}^{neutro}}\Bigg)](https://upload.wikimedia.org/math/5/f/5/5f560d45aff10b38769528e4e1aa6311.png)
Además, log D es pH dependiente, por tanto, se debe especificar a qué pH se ha medido log D. De particular interés es el cálculo de log D para pH = 7.4 (el pH fisiológico del suero sanguíneo. Para un compuesto no ionizable, log P = log D para cualquier pH.
Métodos de medida del coeficiente de reparto, logP
Existen dos procedimientos distintos, si bien antes de llevar a cabo estos procedimientos experimentales se ha de tener una estimación previa del coeficiente de reparto, que se calcula como cociente entre las solubilidades por separado de la sustancia en n-octanol y en agua :
![P_{estimado} = \frac{\big[soluto\big]_{n-octanol} \quad de \ saturaci\acute{o}n}{\big[soluto\big]_{agua}\quad de \ saturaci\acute{o}n}](https://upload.wikimedia.org/math/9/2/9/929c4a0063b0c8a33d671d12756d7c53.png)
- El método del frasco de agitación , para valores de logP comprendidos entre -2 y 4. Es aplicable sólo a sustancias puras solubles en agua y n-octanol. No se puede aplicar a productos tensoactivos. En este caso se da su valor calculado o una estimación ).
- La cromatografía líquida de alta resolución (CLAR), para valores de logP comprendidos entre 0 y 6. Es menos sensible a la presencia de impurezas. Las impurezas dificultan, a veces, la interpretación de los resultados.
Aplicaciones
Farmacología
Farmacocinética
En el contexto de la farmacocinética (lo que le ocurre a un fármaco en el cuerpo), el coeficiente de distribución tiene una gran influencia sobre las propiedades ADME(Absorción, Distribución, Metabolismo, y Excreción) del fármaco. Por consiguiente, el carácter hidrófobo de un compuesto (medido por su coeficiente de distribución ) es un determinante principal de cómo es su potencial uso como fármaco. Más específicamente, para que un fármaco se absorba por vía oral, normalmente debe pasar primero a través de una capa lipídica en el epitelio intestinal (un proceso llamado transporte transcelular). Para un transporte eficiente, el fármaco debe ser suficientemente hidrófobo para que entre en la capa lipídica, pero no tan hidrófobo como para que, una vez en la bicapa, no se difunda de nuevo fuera de la bicapa.2Asimismo, el carácter hidrófobo juega un papel importante en la determinación del lugar donde se distribuyen los fármacos en el cuerpo después de la adsorción y, como consecuencia, de la velocidad a la que serán metabolizados y excretados.
Farmacodinámica
En el contexto de la [[Farmacodinámica] (lo que un fármaco hace al cuerpo), el efecto hidrofóbico es la mayor fuerza conductora para la unión de los fármacos a susreceptores diana.3 4 Por otra parte, los fármacos hidrófobos tienden a ser más tóxicos porque, en general, son retenidos durante un tiempo más largo, tienen una distribución más amplia dentro del cuerpo (por ejemplo, intracelular), son algo menos selectivos en su unión con las proteínas, y finalmente son a menudo ampliamente metabolizados. En algunos casos,los metabolitos pueden ser químicamente reactivos. Asimismo se aconseja que el fármaco sea tan hidrófilo como sea posible mientras retenga la suficiente afinidad de enlace a las proteínas terapéuticas diana. 5 Por tanto, el coeficiente de distribución ideal para un fármaco es habitualmente intermedio (ni demasiado hidrofóbico ni demasiado hidrofílico).
Productos de consumo
Otras muchas industrias tienen en cuenta los coeficientes de distribución, por ejemplo, en la formulación de maquillajes, cremas tópicas, tintes, colorantes para el pelo y muchos otros productos de consumo.
Productos agroquímicos
Los insecticidas y herbicidas hidrofóbicos tienden a ser más activos. Por otra parte, los productos agroquímicos hidrofóbicos tienen vidas medias más largas y por tanto muestran un riesgo aumentado de impacto negativo sobre el medio ambiente.
Metalurgia
En metalurgia, el coeficiente de reparto es un factor importante en la determinación del modo en que las impurezas se distribuyen entre el metal fundido y el metal solidificado. Es un parámetro crítico para el método de purificación de sustancias de fusión por zonas, y determina el grado de efectividad con que puede eliminarse una impureza mediante la solidificación direccional, descrita por la ecuación de Scheil.
Medio ambiente
El carácter hidrófobo de un compuesto puede dar a los científicos una indicación de la facilidad con la que un compuesto podría ser arrastrado hasta las aguas subterráneas y contaminar las vías fluviales.6 Los coeficientes de distribución pueden medirse o ser predecidos para compuestos que actualmente causan problemas o con la previsión de evaluar las modificaciones estructurales necesarias para hacer un compuesto más en la fase de investigación. En el campo de la hidrogeología, el coeficiente de reparto octanol-agua, o Kow, se usa para predecir y modelar la migración de los compuestos orgánicos hidrofóbicos en el suelo y las aguas subterráneas.
Medida del coeficiente de reparto
Método del frasco (o tubo) de agitación

El método clásico y más fiable de determinación de log P es el método del frasco de agitación, que consiste en la disolución de algo del soluto en cuestión en un volumen de octanol y agua, y la posterior medida de la concentración del soluto en cada disolvente. El método más común para medir la distribución del soluto es por espectroscopia UV/VIS. Existen algunos pros y contras en este método:
Pros:
- Es el método más preciso.
- Preciso para el rango más amplio de solutos (aplicable a compuestos neutros y cargados)
- La estructura química no tiene que ser conocida de antemano.
Contras:
- Consumo de tiempo (>30 minutos por muestra)
- El octanol y el agua deben ser previamente mezclados y equilibrados (se tarda al menos 24 horas en equilibrar)
- Se debe alcanzar una solubilidad completa, y puede ser difícil detectar pequeñas cantidades de material sin disolver.
- La respuesta concentración vs. respuesta UV-Vis debe ser lineal en todo el rango de concentración del soluto. (Véase la ley de Lambert-Beer)
- Si el compuesto es extremadamente lipofílico o hidrofílico, la concentración en una de las fases será extremadamente pequeña, y por tanto difícil de cuantificar.
- En relación con los métodos cromatográficos, se requieren grandes cantidades de material.
Como alternativa a la espectroscopia UV/VIS se pueden usar otros métodos para medir la distribución, uno de los mejores consiste en el uso de un trazador radioactivolibre de arrastre. En este método (que resulta muy adecuado para el estudio de la extracción de metales), una cantidad conocida de material radiactivo se añade a una de las fases. Luego se ponen las dos fases en contacto y se mezclan hasta que se alcance el equilibrio. Luego se separan las dos fases y se mide la radiactividad de las dos fases. Usando un detector dispersivo de energía (como un detector de germanio de alta pureza) se permite el uso de varios metales radioactivos diferentes de una vez, mientras que los detectores gamma más sencillos sólo permiten que se use un elemento radiactivo en la muestra.
Si los volúmenes de ambas fases coinciden, entonces los cálculos son muy fáciles.
Para un soluto hipotético (S)
 y también
y también ![\quad \quad \mathbf{D} \quad \acute{o} \quad \mathbf{P} \ = \ \frac {[S_{org\acute{a}nica}]} {[S_{acuosa}]} \,](https://upload.wikimedia.org/math/e/d/c/edcc930ad9aefd643866f6968f15dac6.png)
En tal experimento, usando un radioisótopo libre de arrastre, la carga de disolvente es muy pequeña, por consiguiente los resultados son diferentes de los que se obtendrían cuando la concentración de soluto es muy alta. Una desventaja del experimento del radioisótopo libre de arrastre es que el soluto puede absorberse sobre las superficies del instrumental de vidio (o plástico) o en la superficie de separación entre las dos fases. Para evitar esto,debemos calcular el balance de masas.
Debe cumplirse que
Radiactividad de la fase orgánica + Radiactividad de la fase acuosa = Radiactividad inicial de la fase que contiene el radiotrazador.
Para metales no radiactivos, en algunos casos es posible usar ICP-MS o ICP-AES. Desgraciadamente los métodos ICP sufren frecuentes interferencias por lo que no se aplica la espectroscopia gamma y, por consiguiente, el uso de radiotrazadores (contados por espectroscopia de rayos gamma) es, con frecuencia, más sencillo.
Determinación por Cromatografía líquida de alta resolución (Método CLAR)
Un método más rápido para determinar log P emplea la Cromatografía líquida de alta resolución (Método CLAR). El log P de un soluto se puede determinar porcorrelación de su tiempo de retención con compuestos similares de log P conocido.7
Pros:
- Es un método rápido de determinación (5-20 minutos por muestra)
Contras:
- La estructura química del soluto debe ser conocida de antemano.
- Ya que el valor de log P se determina por regresión lineal, varios compuestos con estructuras similares deben tenar valores de log P conocidos.
- Diferentes familias de compuestos químicos tendrán diferentes coeficientes de correlación; las comparaciones entre distintas familias no son significativas.
Métodos electroquímicos
En el pasado reciente, se han realizado algunos experimentos utilizando interfaces líquidas polarizadas para examinar la termodinámica y la cinética de la transferencia de especies químicas cargadas de una fase a otra. Existen dos métodos principales:
- ITIES, (en inglés, Interfaces between two immiscible electrolyte solutions), interfaces entre dos disoluciones de electrolitos inmiscibles8 which for example has been used at Ecole Polytechnique Fédérale de Lausanne. [1]
- Experimentos de reacciones electroquímicas en gotitas que han sido desarrollados por Alan Bond, Frank Marken[2] y también por el equipo de la École Polytechnique Fédérale de Lausanne. En este caso una reacción en una interfaz triple entre un sólido conductor, gotitas de una fase líquida activa redox y una solución de electrólito se ha empleado para determinar la energía requerida para transferir especies cargadas a través de la interfaz.9
Predicción de valores de logP
Los algoritmos del modelo de relación cuantitativa estructura-propiedades (QSPR,por sus sigls en inglés, Quantitative Structure-Property Relationship) calculan log Pde varias maneras diferentes:
- Predicción basada en estructuras atómicas (contribución atómica; AlogP, MlogP, etc.)
- El método más simple para predecir logP consiste en parametrizar las contribuciones de varios átomos al coeficiente de partición molecular global.
usando ajuste por mínimos cuadrados limitado a un conjunto de compuestos de entrenamiento con coeficientes de partición medidos experimentalmente.10 11 12 Para obtener correlaciones razonables, los elementos químicos más frecuentes contenidos en las moléculas de fármaco (hidrógeno, carbono, oxígeno, azufre, nitrógeno, y halógenos) se dividen en varios tipos de átomos diferentes dependiendo del entorno del átomo dentro de la molécula. Mientras este método es generalmente el menos preciso, la ventaja es que es el más general, siendo capaz de suministrar al menos una estimación aproximada para un amplio conjunto de moléculas.
- Predicción basada en el fragmento (contribución del grupo ; ClogP, etc.)
- Se ha demostrado que el valor de log P de un compuesto se puede determinar por la suma de sus fragmentos moleculares que no se solapan (definidos como uno o más átomos enlazados covalentemente a otros dentro de la molécula). Los valores de log P fragmentarios han sido determinados por un método estadístico análogo a los métodos atómicos (ajuste pormínimos cuadrados de un conjunto de entrenamientol). Además, se incluyen correcciones tipo Hammett para tener en cuenta los efectos electrónico y estérico. Este método da en general mejores resultados que los basados en los átomos, pero no puede usarse para predecir coeficientes de reparto de moléculas que contienen grupos funcionales inusuales porque el método no ha sido todavía bien parametrizado (lo más probable es que sea debido a la falta de datos experimentales para moléculas que contienen tales grupos funcionales).13 14
- Predicción basada en minería de datos
- Una predicción típica basada en minería de datos utiliza máquinas de vectores de soporte,15 árboles de decisión, or redes neuronales.16 Este método consigue habitualmente un éxito notable al calcular los valores de log P cuando se emplea con compuestos que tienen estructuras químicas similares y valores de log Pconocidos.
- Predicción basada en minería de moléculas
- Las aproximaciones de minería de moléculas aplican una predicción basada en matrices de similaridad o de adyacencia o un esquema de fragmentación automática en subestructuras moleculares. Adicionalmente también existen aproximaciones que emplean búsquedas del subgrafo común máximo o del kernel de moléculas.
- Estimación de log D (para un pH dado) a partir de log P y pKa:17
- Expresiones exactas:
- Aproximaciones para compuestos ampliamente ionizados:
- Aproximaciones para compuestos ampliamente no-ionizados:
- Predicción de pKa
- Para la predicción de pKa que a su vez puede usarse para estimar log D, se han aplicado frecuentemente las ecuaciones tipo Hammett.18 Ver19 para la revisión más reciente de los métodos más modernos.
![log\ D_{\acute{a}cidos} = log\ P + log\Bigg[\frac{1}{(1+10^{pH-pK_a})}\Bigg]](https://upload.wikimedia.org/math/9/5/e/95ec27680c4e563fe56ca346601378ef.png)
![log\ D_{bases} = log\ P + log\Bigg[\frac{1}{(1+10^{pK_a-pH})}\Bigg]](https://upload.wikimedia.org/math/7/0/b/70b14db31996c707a40236716853f0c9.png)



Limitaciones
El logaritmo del coeficiente de reparto, Log P, no es un determinante preciso del carácter lipofílico de compuestos ionizables porque sólo describe correctamente los coeficientes de reparto de moléculas neutras (no cargadas). Tomando el ejemplo del descubrimiento de fármacos vemos como las limitaciones de log P pueden afectar a la investigación. Ya que la mayoría de los fármacos, (aproximadamente el 80%) son ionizables, log P no es un predictor apropiado del comportamiento de un compuesto en los entornos de pH cambiantes del cuerpo. El coeficiente de distribución (Log D) es el descriptor correcto para sistemas ionizables.
Alternativamente, se puede usar un coeficiente de reparto aparente que se define como sigue:  .
.
.
Claramente, si el fármaco está 100% sin ionizar entonces  .
.
.
depuración, separación, o aclaramiento (CL, del inglés clearance) de una sustancia es el inverso del tiempo constanteque describe su índice de eliminación del cuerpo dividido por su volumen de distribución (o total de agua corporal).
El aclaramiento, puede determinarse de una forma global (aclaramiento sistémico) o de forma individualizada para cada vía (aclaramiento hepático, renal, etc.). La ecuación que recoge este concepto sería:
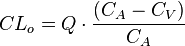
En donde  es el aclaramiento del órgano,
es el aclaramiento del órgano,  la concentración plasmática en sangre arterial,
la concentración plasmática en sangre arterial,  la concentración plasmática en sangre venosa y
la concentración plasmática en sangre venosa y 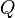 el flujo sanguíneo del órgano.
el flujo sanguíneo del órgano.
es el aclaramiento del órgano, la concentración plasmática en sangre arterial, la concentración plasmática en sangre venosa y el flujo sanguíneo del órgano.
Es usado en el área de farmacología, para referirse a la capacidad que tiene un órgano para eliminar un fármaco. Se expresa mediante el número de mililitros de plasma que se depura de una sustancia (fármaco) determinada por minuto. La expulsión del fármaco puede ser consecuencia de procesos que tienen lugar en riñones, hígado y otros órganos.
El aclaramiento renal es usado también para evaluar algunos aspectos de la función renal.
Depuración
El concepto de depuración o aclaramiento ("clearance") implica esencialmente el proceso de eliminación renal de una substancia. Sin embargo, este concepto se ha extendido para describir la eliminación por o desde órganos como el hígado y otros de menor importancia como el estómago, pulmones, etc. El concepto de depuración se emplea ampliamente hoy día en farmacocinética y terapéutica para determinar la vida media biológica de fármacos, para predecir el estado estacionario de un fármaco en la sangre o el plasma durante su administración constante, para evaluar o predecir la biodisponibilidad de un fármaco administrado por diferentes vías y para medir la función de un órgano.
La depuración de un órgano se define como el volumen del medio de perfusión que es purificado de un fármaco por ese órgano en la unidad de tiempo. La depuraci6ntiene, por lo tanto, unidades de volumen por el tiempo reciproco
La depuración es una propiedad aditiva: su valor total es la suma de la depuración de cada uno de los órganos eliminadores.
La mayoría de los fármacos o sus metabolitos se eliminan a través del riñón. La depuración renal se refiere al volumen de plasma que es depurado del fármaco en la unidad de tiempo por el riñón. Algunas substancias son secretadas en alto grado por los túbulos renales de modo que el plasma es completamente depurado en una sola pasada por el riñón. Tales substancias sirven como una medida del flujo plasmático renal. El ácido p-aminohipúrico y la penicilina se aproximan a este ideal; su depuración es de alrededor de 650 ml/min. Como el volumen plasmático es casi la mitad del volumen sanguíneo, el flujo sanguíneo renal a través de ambos riñones es cercano a los 1.300 ml/min medido mediante el ácido p-aminohipúrico.
El carbohidrato inulina, que no se une apreciablemente a las proteínas plasmáticas, filtra a través del glomérulo y no es reabsorbido ni secretado por los túbulos. Sudepuraci6nes, por esta característica, una medida de la velocidad de filtración glomerular. Esta, en el individuo normal, es de alrededor de 130 ml/min. La depuraci6nde la glucosa, en condiciones normales, es nula ya que se reabsorbe completamente a nivel tubular. La urea en el filtrado glomerular difunde fuera de los túbulos a medida que suconcentración aumenta por reducción progresiva del volumen del filtrado a causa de la reabsorción de agua. La depuración de urea endogena ha sido con frecuencia empleada como una medida de la función renal.
La determinación dela depuraci6ndeurea está sujeta a varios errores a causa de su reabsorción variable por los túbulos renales y de factores exógenos que pueden afectar el nivel plasmático de urea. Las dietas proteicas pueden ocasionar errores, muchas veces de consideración; los estados hemorrágicos intestinales, los vómitos y la deshidratación se citan también como causas frecuentes de errores en esta determinación. Por tal motivo, la determinación más exacta es la de la depuración de inulina que, como ya hemos señalado, filtra por el glomérulo sin ser reabsorbida ni secretada por los túbulos. Esta determinación es compleja desde el punto de vista analítico, por lo que se prefiere emplear la creatinina que presenta menos complicaciones. Esta substancia también filtra libremente por el glomérulo. Sin embargo, una pequeña fracción es también secretada por los túbulos renales, lo cual puede ocasionar una sobreestimación de la verdadera velocidad de filtración glomerular. Esto sucede especialmente cuando esta velocidad desciende a niveles inferiores a 30 ml/min o menos. Por esta razón, en pacientes en que la depuración de creatinina es de 10 ml/min o menos, se considera que la velocidad de filtración glomerular es prácticamente nula.
La depuración renalpuede obtenerse a partir de datos de excreción renal, ya que:
donde dE/dt es la velocidad de excreción renal del fármaco y C la concentración plasmática medida en la mitad del intervalo de tiempo en que se obtienen las muestras de orina. En un gráfico de dE/dt en función de C, la depuración renal está determinada por la pendiente de la recta obtenida. Otra manera de calcular la depuración renal consiste en determinar la cantidad total del fármaco excretado por la orina y en dividir esta cantidad por el área total bajo la curva de concentración plasmática en función del tiempo:
También la depuración hepática es importante en farmacocinética. El hígado es el principal órgano eliminador de fármacos. Situado entre el tracto gastrointestinal y la circulación general, recibe la mayor parte de la sangre que proviene de dicho tracto y, por lo tanto, los fármacos absorbidos allí pasarán primero por el hígado, Por la gran capacidad metabolizante de este órgano, puede reducirse la disponibilidad de fármacos administrados por vía oral, es decir, la fracción de la dosis oral que llega intacta a la circulación sistemática. Esta eliminación hepática presistémica de un fármaco es lo que se llama el efecto del primer paso y es particularmente importante en los fármacos que son "extraídos" en alto grado por el hígado, como el propranolol y el propoxifeno.
Una expresión ampliamente utilizada en la actualidad es la de "relación de extracción hepática" o "porcentaje de extracción hepática", que es el porcentaje de fármaco irreversiblemente metabolizado a cada paso por el hígado:
donde
= fracción de la dosis metabolizada, y
= constante que expresa la relación entre la concentración sanguínea y plasmática.


La eliminación hepática puede estar limitada por el flujo sanguíneo y por la capacidad intrínseca del órgano para eliminar fármacos. En algunos fármacos, la relación de extracción hepática es cercana a la unidad, como es el caso del propranolol, la lidocaína y el propoxifeno. El hígado tiene una gran capacidad intrínseca de metabolizar estos fármacos y en ellos la depuración hepática llega a ser prácticamente igual al flujo sanguíneo hepático: 1, 34 1/h por Kg.
Por otro lado, hay fármacos cuya relación de extracción hepática se aproxima a cero, como sucede con la antipirina y la warfarina. Aquí, la limitante no es el flujo sanguíneo sino la escasa o nula capacidad intrínseca del hígado o de las enzimas hepáticas para metabolizar estos fármacos.
También es preciso considerar el efecto de la unión a proteínas, ya que la velocidad del metabolismo depende del fármaco libre. La depuración de un fármaco escasamente extraído es sensible a los cambios de unión a proteínas de la sangre, por ejemplo, la warfarina, en la cual la depuración es directamente proporcional a la fracción del fármaco no unido alas proteínas del plasma.
La depuración total de un fármaco es el resultado de la eliminación de éste por todos los mecanismos eliminatorios del organismo: excreción renal, hepática, pulmonar, metabólico, etc., y, por lo tanto, está relacionado con la constante de velocidad de eliminación total, la cual, como se ha señalado antes, es la suma de todas las constantes de velocidad de eliminación individuales de los mecanismos que se encargan de esta función.
La ecuación que relaciona la depuración total con la constante de velocidad de eliminación total es:
También puede expresarse la depuración totalenfunci6n de la vida media biológica o tiempo medio de semieliminación:

No hay comentarios:
Publicar un comentario