Farmacocinética
La distribución de los fármacos puede definirse, entre otras formas, como la llegada y disposición de un fármaco en los diferentes tejidos del organismo. Es un proceso muy importante, toda vez que, según su naturaleza, cada tejido puede recibir cantidades diferentes del fármaco, el cual, además, pasará allí tiempos variables.1
A la hora de hablar de la distribución, habrá que tener en cuenta los conceptos sobre compartimentación del organismo vistos en el apartado de Modelos farmacocinéticos.
Factores que afectan la distribución
Son múltiples, pero siguiendo a Pascuzzo, los más importantes son los tres siguientes: los volúmenes físicos del organismo, la tasa de extracción y la unión a proteínas plasmáticas y, o, tisulares.
Volúmenes físicos del organismo
Este concepto está relacionado con la multicompartimentalización. Considerando los fármacos como solutos, los distintos tejidos con especificidad del organismo van a actuar como los solventes que darán pie a las diferentes concentraciones del fármaco. Así, dependiendo de la naturaleza química de éste, habrá una especial predisposición de las sustancias liposolubles por la grasa corporal o de las hidrosolubles por el líquido extracelular. Este volumen de distribución (Vd) de un fármaco en el organismo es tan sólo aparente, pues conceptualmente se trataría del volumen necesario para contener de forma homogénea en todo el organismo una cantidad determinada de fármaco, que viene dada por el nivel de la concentración del mismo en el plasma. Desde el punto de vista físico el Vd viene determinado por la siguiente fórmula: 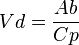 en donde
en donde 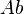 es la cantidad total de fármaco en el cuerpo y
es la cantidad total de fármaco en el cuerpo y  la concentración plasmática del mismo.
la concentración plasmática del mismo.
en donde es la cantidad total de fármaco en el cuerpo y la concentración plasmática del mismo.
Siendo la conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre  y la es una relación de proporcionalidad inversa. Es decir, que a mayor menor y viceversa. O lo que es lo mismo, que los factores que aumenten la disminuirán el valor del . Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
y la es una relación de proporcionalidad inversa. Es decir, que a mayor menor y viceversa. O lo que es lo mismo, que los factores que aumenten la disminuirán el valor del . Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre y la es una relación de proporcionalidad inversa. Es decir, que a mayor menor y viceversa. O lo que es lo mismo, que los factores que aumenten la disminuirán el valor del . Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
Aplicando a esta fórmula los conceptos aprendidos en el apartado de la biodisponibilidad, podemos calcular la cantidad de fármaco a administrar para conseguir una determinada concentración de fármaco en el organismo (dosis de carga):

Este concepto tiene interés clínico, pues a veces necesitamos alcanzar una determinada concentración de fármaco que sabemos es la óptima para que realice sus efectos en el organismo (caso de la digitalización de un paciente).
Tasa de extracción
Se refiere a la proporción del fármaco que es retirado de la circulación por cada órgano, una vez que el flujo sanguíneo lo haya hecho pasar a través de dicho órgano.1Este nuevo concepto integra otros anteriores, ya que la tasa de extracción va a depender de distintos factores:
- Características del fármaco, entre ellas su pKa.
- Redistribución tisular: En algunos fármacos se produce una distribución rápida e intensa en determinados tejidos, hasta llegar al equilibrio con la concentración plasmática. Sin embargo otros tejidos más lentos continúan retirando fármaco del plasma, con lo que la concentración en el primer tejido queda por encima de la plasmática y por tanto sale fármaco del tejido hacia el plasma. Este fenómeno se sigue sucediendo durante un tiempo hasta alcanzar el equilibrio definitivo. Se obtiene pòr tanto dos concentraciones del fármaco en el tejido más sensible: una inicial más elevada y otra posterior consecuencia de la redistribución tisular.
- Diferencial de concentración con los tejidos.
- Superficie de intercambio.
- Presencia de barreras naturales. Son obstáculos a la difusión similares a las encontradas en la absorción. Las más interesantes son:
- Permeabilidad de los lechos capilares, que no es igual en todos los tejidos.
- Barrera hematoencefálica: está localizada entre el plasma sanguíneo de los vasos cerebrales y el espacio extracelular del encéfalo. Dificulta la llegada de fármacos al mismo.
- Barrera placentaria: en la mujer embarazada, evita la llegada de gran cantidad de fármacos al feto, que pudieran ser tóxicos para el mismo.
Unión a proteínas plasmáticas
Algunos fármacos tienen la capacidad de unirse a distintos tipos de proteínas vehiculizadas en el plasma sanguíneo. Esto es de gran importancia dado que, como sabemos, sólo el fármaco que se encuentra diluido en el plasma será capaz de pasar a los tejidos. De esta manera la unión del fármaco a las proteínas plasmáticas actúa como un reservorio del mismo dentro del organismo y disminuye las concentraciones finales en los tejidos. La unión de fármacos y proteínas es poco específica y usualmente lábil y reversible, generalmente a través de enlaces iónicos, puentes de hidrógeno, fuerzas de Van der Waals y, con menos frecuencia, enlaces covalentes. Esto implica que un fármaco puede ser desplazado de su unión a la proteína por otra sustancia (u otro fármaco) y que en todo caso, la unión está sujeta a saturación. También, existe un equilibrio entre el fármaco libre en el plasma y el unido a proteínas, por lo que la proporción de fármaco unido a las mismas es estable, independientemente de su cantidad total en el plasma.
Por estudios realizados in vitro en condiciones ideales, el equilibrio entre la concentraciones plasmática y tisular del fármaco sólo se ve alterado de forma significativa con índices de fijación a proteínas plasmáticas superiores al 90%. A partir de estos niveles se produce un "secuestro" del fármaco que disminuye su presencia en los tejidos por debajo del 50% del total. Esto es importante a la hora de considerar las interacciones farmacológicas: un fármaco con un índice de fijación a proteínas plasmáticas inferior al 90%, si es desplazado de su unión a las proteínas por otro fármaco no va a aumentar significativamente su presencia en los tejidos. Por el contrario, con índices de unión a proteínas plasmáticas superiores al 95%, pequeños desplazamientos pueden originar importantes modificaciones de la concentración tisular y, por tanto, mayor riesgo de toxicidad por exceso de su efecto en los tejidos.
De las proteínas plasmáticas quizás las de más interés sean las albúminas, por su cantidad y su capacidad para unirse a otras sustancias. Otras proteínas de interés son las glicoproteínas, las lipoproteínas y en menor medida las globulinas.
Como podrá comprenderse, situaciones clínicas que supongan modificación de los niveles de proteínas plasmáticas (por ejemplo hipoalbuminemias secundarias a procesos renales) pueden tener transcendencia en el efecto y toxicidad de un fármaco que presente índices de unión a proteínas plasmáticas superiores al 90% (ó 0,9).


En farmacología se entiende por eliminación o excreción de un fármaco a los procesos por los cuales los fármacos son eliminados del organismo, bien inalterados (moléculas de la fracción libre) o bien modificados como metabolitos a través de distintas vías. El riñón es el principal órgano excretor, aunque existen otros, como elhígado, la piel, los pulmones o estructuras glandulares, como las glándulas salivales y lagrimales. Estos órganos o estructuras utilizan vías determinadas para expulsar el fármaco del cuerpo, que reciben el nombre de vías de eliminación:
En lo que respecta al riñón, los fármacos son excretados por filtración glomerular y por secreción tubular activa siguiendo los mismos pasos y mecanismos de los productos del metabolismo intermedio. Así, las drogas que filtran por el glomérulo sufren también los procesos de la reabsorción tubular pasiva. Por filtración glomerular solo se eliminan las drogas o los metabolitos no ligados a las proteicas plasmáticas (fracción libre), y muchos otros (como los ácidos orgánicos) son secretados activamente. En los túbulos proximal y distal las formas no ionizadas de ácidos o bases débiles son reabsorbidas pasiva y activamente. Cuando el fluido tubular se hace más alcalino, los ácidos débiles se excretan más fácilmente y esto disminuye la reabsorción pasiva. Lo inverso ocurre con las bases débiles. Por eso en algunas intoxicaciones puede incrementarse la eliminación del fármaco tóxico, alcalinizando la orina y forzando la diuresis.
En otras ocasiones los fármacos son eliminados en la bilis con la que llegan hasta el intestino. Allí se unen a la fracción no absorbida del fármaco y se eliminan con las heces o bien pueden sufrir un nuevo proceso de absorción y ser eliminados finalmente por el riñón.
Parámetros farmacocinéticos de la excreción
La farmacocinética estudia la forma y velocidad de depuración de los fármacos y sus metabolitos por los distintos órganos excretores, en relación con las concentraciones plasmáticas del fármaco. Para ello precisa de la definición operativa de algunos conceptos relativos a la excreción.
Biodisponibilidad
A efectos prácticos, podemos definir la biodisponibilidad de un fármaco como la fracción del mismo que alcanza la circulación sistémica del paciente. O dicho de otra manera, el porcentaje de fármaco que aparece en plasma. Desde este prisma, la administración de un fármaco por vía intravenosa presentaría la mayor biodisponibilidad posible, por lo que se considera la unidad (o el 100%). A partir de aquí, la biodisponibilidad se calcula comparando la vía a estudiar con respecto a la vía intravenosa (biodisponibilidad absoluta) o a un valor estándar de otras presentaciones del fármaco en estudio (biodisponibilidad relativa).
![B_A = \frac{[ABC]_P . D_{IV}}{[ABC]_{IV} . D_P}](https://upload.wikimedia.org/math/5/c/a/5ca3c5293ac76c5cc118262203196262.png)
.
![\mathit B_R = \frac{[ABC]_{A} . dosis_{B}}{[ABC]_{B} . dosis_{A}}](https://upload.wikimedia.org/math/8/9/1/89142147383556850b8ddeefa9d174b1.png)
Conocida la biodisponibilidad de un fármaco, podremos calcular qué modificaciones hay que realizar en su posología para alcanzar los niveles sanguíneos deseados. La biodisponibilidad es pues una razón matemática individal para cada fármaco que actúa sobre la dosis administrada. Mediante la fórmula 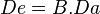 (en donde Dees la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
(en donde Dees la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
(en donde Dees la dosis eficaz, B la biodisponibilidad y Da la dosis administrada) podemos calcular la cantidad de fármaco en plasma que realmente tiene capacidad para realizar su efecto.
Así, si tenemos un fármaco cuya biodisponibilidad es de 0.8 (o del 80%) y se administra una dosis de 100 mg, la ecuación se resolvería:
De = 0,8 x 100 mg = 80 mg
Este concepto depende de otra serie de factores inherentes a cada fármaco, como son:1
- Forma galénica
- Forma química
- Vía de administración
- Estabilidad
- Metabolización
Estos conceptos, que pueden verse detalladamente en el artículo principal del epígrafe, pueden cuantificarse matemáticamente y a su vez ser integrados para obtener un ecuación matemática de los mismos:

,
donde Q sería la constante de pureza del fármaco.1

siendo  la velocidad de administración del fármaco y
la velocidad de administración del fármaco y  la constante que representa la velocidad a la que el fármaco absorbido alcanza la circulación sistémica.
la constante que representa la velocidad a la que el fármaco absorbido alcanza la circulación sistémica.
la velocidad de administración del fármaco y la constante que representa la velocidad a la que el fármaco absorbido alcanza la circulación sistémica.
Finalmente, por la ecuación de Henderson-Hasselbach, y sabiendo el  del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
del fármaco (pH al cual presenta equilibrio entre sus moléculas ionizadas y no ionizadas), podemos calcular la cantidad de fármaco no ionizado, y, por tanto, la cantidad de fármaco objeto de la absorción:
Cuando dos fármacos tienen la misma biodisponibilidad se dice que son equivalentes biológicos o bioequivalentes. Este concepto de bioequivalencia es importante porque en la actualidad es la vara de medir para la autorización de los medicamentos genéricos en numerosos países.
Volumen aparente de distribución
Este concepto está relacionado con la teoría de la multicompartimentalización. Considerando los fármacos como solutos, los distintos tejidos con especificidad del organismo van a actuar como los solventes que darán pie a las diferentes concentraciones del fármaco. Así, dependiendo de la naturaleza química de éste, habrá una especial predisposición de las sustancias liposolubles por la grasa corporal o de las hidrosolubles por el líquido extracelular. Este volumen de distribución (Vd) de un fármaco en el organismo es tan sólo aparente, pues conceptualmente se trataría del volumen necesario para contener de forma homogénea en todo el organismo una cantidad determinada de fármaco, que viene dada por el nivel de la concentración del mismo en el plasma. Desde el punto de vista físico el Vd viene determinado por la siguiente fórmula: en donde es la cantidad total de fármaco en el cuerpo y la concentración plasmática del mismo.
en donde es la cantidad total de fármaco en el cuerpo y la concentración plasmática del mismo.
Siendo la conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre y la es una relación de proporcionalidad inversa. Es decir, que a mayor menor y viceversa. O lo que es lo mismo, que los factores que aumenten la disminuirán el valor del . Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
conocida, pues es equivalente a la dosis de fármaco administrada, la fórmula nos indica que la relación existente entre y la es una relación de proporcionalidad inversa. Es decir, que a mayor menor y viceversa. O lo que es lo mismo, que los factores que aumenten la disminuirán el valor del . Esto nos pone sobre la pista de la importancia del conocimiento de las concentraciones plasmáticas del fármaco y de los factores que lo modifican.
Aplicando a esta fórmula los conceptos aprendidos en el apartado de la biodisponibilidad, podemos calcular la cantidad de fármaco a administrar para conseguir una determinada concentración de fármaco en el organismo (dosis de carga):
Este concepto tiene interés clínico, pues a veces necesitamos alcanzar una determinada concentración de fármaco que sabemos es la óptima para que realice sus efectos en el organismo (caso de la digitalización de un paciente).
Vida media
La vida media plasmática o vida media de eliminación es una constante que indica el tiempo necesario para eliminar el 50% del fármaco del organismo,o bien el tiempo que tarda la concentración plasmática del fármaco en reducirse a la mitad de sus niveles máximos.
Aclaramiento
Al medir la concentración plasmática de un fármaco antes de pasar por un órgano (sangre arterial) y después de haber pasado por él (sangre venosa) si se encuentra una diferencia de concentraciones se puede deducir que el órgano ha eliminado una parte del fármaco, aclarando la concentración del mismo. Desde esta óptica, se considera el aclaramiento como el volumen plasmático libre totalmente de fármaco por unidad de tiempo, por lo que se mide en unidades de volumen por unidades de tiempo. El aclaramiento, puede determinarse de una forma global (aclaramiento sistémico) o de forma individualizada para cada vía (aclaramiento hepático, renal, etc.). La ecuación que recoge este concepto sería:
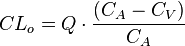
En donde  es el aclaramiento del órgano,
es el aclaramiento del órgano,  la concentración plasmática en sangre arterial,
la concentración plasmática en sangre arterial,  la concentración plasmática en sangre venosa y
la concentración plasmática en sangre venosa y 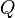 el flujo sanguíneo del órgano.
el flujo sanguíneo del órgano.
es el aclaramiento del órgano, la concentración plasmática en sangre arterial, la concentración plasmática en sangre venosa y el flujo sanguíneo del órgano.
Es fácil comprender que cada órgano tendrá sus condicionantes del aclaramiento, en función de su mecanismo de acción para realizar la depuración. En lo que respecta al aclaramiento renal, viene determinado por factores como el grado de unión a proteínas plasmáticas del fármaco (sólo se filtra el fármaco libre), saturación de los transportadores (la secreción activa depende de proteínas transportadoras, que son saturables), o el número de nefronas funcionantes (de donde la importancia de situaciones como la insuficiencia renal).
En el caso del hígado, el aclaramiento hepático es fruto del metabolismo y por tanto está determinado por los factores que alteran el mismo así como por la cantidad de hepatocitos funcionantes, lo que justifica la importancia clínica de la insuficiencia hepática.
Estado de equilibrio
El estado de equilibrio o concentración estable es aquel en el que los aportes plasmáticos de fármaco se equilibran con la eliminación del mismo. Es fundamental su cálculo para decidir el período entre dosis y la cantidad de fármaco en cada una de ellas, en tratamientos prolongados.
Farmacocinética general: eliminación
Los procesos de eliminación de un fármaco incluyen dos situaciones fisiológicas: la biotransformación y la excreción. La biotransformación ocurre preferentemente en el hígado, pero no exclusivamente, ya que el intestino, la placenta y el pulmón pueden participar de dicho proceso, el que tiene como objetivo la transformación enzimática de cualquier sustancia exógena al organismo en metabolitos hidrosolubles para facilitar la excreción renal, ya que mientras más liposoluble es un fármaco, más tiempo permanecerá en el organismo. Por ejemplo, el insecticida organofosforado DDT es tan liposoluble que permanece en el hígado sin ser metabolizado, por lo que no se elimina.
Un fármaco hidrosoluble se puede filtrar o secretar a nivel del túbulo renal y al llegar a la orina no se reabsorbe, por lo que se elimina. La transformación enzimática en metabolitos hidrosolubles puede originar metabolitos farmacológicamente activos; por ejemplo, el diazepam tiene una vida media de 36 horas, pero además tiene un metabolito activo cuya vida media es de 100 horas, por lo que también se debe metabolizar dicho metabolito a nivel hepático para que cese el efecto del fármaco. Cuando un paciente ingiere una sobredosis aguda de paracetamol, éste destruye las reservas de glutatión hepático, lo que genera la producción de metabolitos toxicológicamente activos que provocan una necrosis hepática aguda. El resto de los fármacos y sus metabolitos son inactivados a nivel hepático (Fig. 1).
Un fármaco hidrosoluble se puede filtrar o secretar a nivel del túbulo renal y al llegar a la orina no se reabsorbe, por lo que se elimina. La transformación enzimática en metabolitos hidrosolubles puede originar metabolitos farmacológicamente activos; por ejemplo, el diazepam tiene una vida media de 36 horas, pero además tiene un metabolito activo cuya vida media es de 100 horas, por lo que también se debe metabolizar dicho metabolito a nivel hepático para que cese el efecto del fármaco. Cuando un paciente ingiere una sobredosis aguda de paracetamol, éste destruye las reservas de glutatión hepático, lo que genera la producción de metabolitos toxicológicamente activos que provocan una necrosis hepática aguda. El resto de los fármacos y sus metabolitos son inactivados a nivel hepático (Fig. 1).
Figura 1. Eliminación de fármacos. Remoción irreversible del fármaco del cuerpo por todas las rutas.
El metabolismo es un proceso secuencial que ocurre en dos fases, donde intervienen dos grupos enzimáticos. Durante el proceso enzimático de fase I los fármacos se convierten en sustancias muy reactivas; aquí el citocromo P-450 hepático juega un rol fundamental. El derivado polar reactivo de la fase I será el sustrato de las enzimas de fase II, en la cual puede experimentar procesos de glucoronidación, acetilación y metilación, además de adición de aminoácidos o glutatión. Cuando el fármaco reactivo se conjuga con una de estas moléculas pierde reactividad y liposolubilidad, es decir, se convierte en una sustancia hidrosoluble que será fácilmente eliminada. Existen fármacos que no se metabolizan, como es el caso de la penicilina, que se excreta del organismo tal cual entra. Los antiinflamatorios no esteroidales (AINES) se excretan en 98% sin alteración y los salicilatos pueden sufrir algún grado de glucoronidación (Fig. 2)
Figura 2. Fases del metabolismo de los fármacos.
El citocromo P-450 es un conjunto de proteínas con actividad enzimática oxidante que se ubican en el retículo endoplasmático liso (microsomas) del hepatocito. Este sistema es inespecífico y fácilmenteinducible, es decir, su actividad aumenta en presencia de una sustancia; es por esto que un paciente fumador requiere cantidades mayores de aminofilina que un paciente no fumador, debido a que sus enzimas hepáticas están inducidas. A su vez, el citocromo P-450 es potencialmente saturable, porque existe una cantidad finita de enzima; esto significa que si se aporta un exceso de sustancia, el sistema se satura. También es fácilmente inhibible; por ejemplo, la eritromicina es capaz de inhibir las enzimas hepáticas que metabolizan el cisapride, fármaco que por sí solo tiene toxicidad cardíaca, motivo por el cual la combinación cisapride-eritromicina no se debe usar, sobre todo en niños, porque puede producir arritmias fatales.
Los factores que determinan la eficiencia del hígado para eliminar fármacos son: la cantidad de fármaco que llega al hígado en la unidad de tiempo, lo que depende del flujo sanguíneo y de la concentración del fármaco en la sangre; la concentración del fármaco libre, es decir, el que no está unido a proteínas plasmáticas; y la actividad de los sistemas enzimáticos implicados en la biotransformación.
Los factores que determinan la eficiencia del hígado para eliminar fármacos son: la cantidad de fármaco que llega al hígado en la unidad de tiempo, lo que depende del flujo sanguíneo y de la concentración del fármaco en la sangre; la concentración del fármaco libre, es decir, el que no está unido a proteínas plasmáticas; y la actividad de los sistemas enzimáticos implicados en la biotransformación.
Excreción renal de fármacos
Existen tres mecanismos de excreción renal de fármacos, que pueden operar solos o en combinación con otros:
Filtración glomerular: es un proceso unidireccional que depende directamente de la fracción libre de la droga. Toda sustancia que llegue al glomérulo será filtrada, siempre y cuando el tamaño molecular no sea demasiado grande o bien, que la fracción de la sustancia que llegue al glomérulo no pueda ser filtrada debido a su unión a las proteínas, porque de esta forma tiene un tamaño molecular mayor. La tasa de filtración glomerular normal es de 125 a 130 mL/min.
Secreción tubular activa: requiere de sistemas transportadores saturables y depende del flujo plasmático renal (valor normal: 425-650 mL/min). En el túbulo proximal, esas sustancias pueden ser secretadas en forma activa hacia el lumen tubular, porque la afinidad de los transportadores tubulares es mayor que la afinidad de las proteínas plasmáticas.
Reabsorción tubular: este proceso puede ser activo o pasivo. Está influido por el pH urinario, ya que las moléculas no ionizadas son liposolubles. Si la sustancia es liposoluble, será reabsorbida en el túbulo proximal prácticamente en 100%; sólo aquéllas sustancias ionizadas e hidrosolubles no serán reabsorbidas y por lo tanto, serán excretadas.
Filtración glomerular: es un proceso unidireccional que depende directamente de la fracción libre de la droga. Toda sustancia que llegue al glomérulo será filtrada, siempre y cuando el tamaño molecular no sea demasiado grande o bien, que la fracción de la sustancia que llegue al glomérulo no pueda ser filtrada debido a su unión a las proteínas, porque de esta forma tiene un tamaño molecular mayor. La tasa de filtración glomerular normal es de 125 a 130 mL/min.
Secreción tubular activa: requiere de sistemas transportadores saturables y depende del flujo plasmático renal (valor normal: 425-650 mL/min). En el túbulo proximal, esas sustancias pueden ser secretadas en forma activa hacia el lumen tubular, porque la afinidad de los transportadores tubulares es mayor que la afinidad de las proteínas plasmáticas.
Reabsorción tubular: este proceso puede ser activo o pasivo. Está influido por el pH urinario, ya que las moléculas no ionizadas son liposolubles. Si la sustancia es liposoluble, será reabsorbida en el túbulo proximal prácticamente en 100%; sólo aquéllas sustancias ionizadas e hidrosolubles no serán reabsorbidas y por lo tanto, serán excretadas.
Clearance
El clearance o depuración es un parámetro farmacocinético descriptivo; consiste en el análisis de la capacidad que tiene el organismo para eliminar un fármaco. El clearence se refiere al volumen de plasma que es procesado, por unidad de tiempo, para eliminar un determinado fármaco. La cantidad de fármaco eliminado es proporcional a la concentración sanguínea del fármaco. Si el clearence es muy alto significa que la capacidad de eliminación del órgano es enorme; si es muy bajo, significa que el órgano no tiene mucha capacidad de eliminar el fármaco, de modo que éste se mantiene por más tiempo en el organismo, tanto en la sangre como en los tejidos. Si el fármaco llega a los tejidos esto influirá en la cantidad de fármaco que existe en el volumen procesado por unidad de tiempo: si hay poco fármaco en ese volumen se podría alterar la velocidad con que se va a excretar, porque aunque tenga una capacidad metabólica muy grande el fármaco se puede quedar en el organismo. Por lo tanto, no se puede estimar que a mayor clearance menor duración y a menor clearance mayor duración del fármaco en el organismo, motivo por el que se debe introducir el concepto devolumen de distribución para compensar los cambios en la concentración plasmática. Elclearance relaciona el volumen de distribución con la duración media del fármaco en el organismo; la concentración plasmática de un fármaco es el reflejo del volumen de distribución que este posee (Fig. 3)
Figura 3. Cálculo del clearance de un fármaco.
En condiciones terapéuticas, y considerando que un órgano es capaz de manejar un volumen fijo de plasma, el porcentaje de fármaco que será eliminado en función del tiempo es siempre constante. Por ejemplo, si existe en el plasma una cantidad x/ml de medicamento, en la siguiente unidad de tiempo habrá 50% menos, es decir, 0,5 x/ml y en la siguiente unidad de tiempo habrá 25%, o 0,25 x/ml, es decir, una disminución de 50% de lo anterior. Por lo tanto, la velocidad de desaparición del fármaco en el organismo es rápida, es decir, con cinética de orden 1 (Fig. 4).
Figura 4. Cinética de orden 1
Cuando el sistema de eliminación está saturado, es decir, cuando existe exceso de fármaco y el sistema tiene que manejar una cantidad fija de fármaco cada vez, en este caso la caída de la concentración plasmática del fármaco es constante, una pequeña cantidad por vez. Si tenemos una concentración plasmática de 500 mg/ml y el sistema metabólico de excreción se encuentra saturado, sólo será capaz de manejar una cantidad constante, por ejemplo 15 mg/hora. A la hora habrá 485 mg/ml; entonces, en 2 horas sólo habrán salido 30 mg, cuando en otras condiciones habrían salido 400. Esto corresponde a una cinética de orden 0, donde el sistema está colapsado. Este tipo de cinética se ve cuando un paciente está intoxicado con Aspirina o con etanol; por este motivo la alcoholemia se extrapola al momento del accidente, ya que se pierde una cantidad pequeña, con una relación lineal (Fig. 5).
Figura 5. Cinética de orden 0.
Vida media de eliminación
La vida media de eliminación es el tiempo que demora en disminuir en 50% la concentración plasmática de un fármaco. Cuando se administra un fármaco, cada vida media se produce un proceso de acumulación, pero ese proceso no es infinito, sino que se establece un equilibrio entre lo que ingresa y lo que egresa, que se denomina estado estacionario, que produce las fluctuaciones plasmáticas terapéuticas en un régimen continuo. Este estado se alcanza después de 4 ó 5 vidas medias. Después de 4 vidas medias se completa 94% de la eliminación y se alcanza 94% del estado estacionario.
Si un paciente con insuficiencia cardiaca necesita tomar digital, para alcanzar el estado estacionario tendría que recibir una dosis diaria durante 7 días; entonces, se debe entregar una dosis de carga para alcanzar el estado estacionario en forma rápida. En cambio, si se administra digoxina cada vida media, en 7 días se alcanza el estado estacionario, donde la cantidad que ingresa del fármaco es igual a la que egresa.
La vida media de eliminación depende del volumen de distribución y del clearence. Si hay un volumen de distribución alto significa que hay mucho fármaco en los tejidos, o sea, poco fármaco disponible para ser eliminado y así la disminución de la concentración plasmática será gradual.; por lo tanto, si el volumen de distribución es alto, la vida media del fármaco será mayor. A mayorclearence, la vida media del fármaco será menor, porque por cada volumen que el órgano procese se eliminará todo el fármaco contenido en ese volumen. Por el contrario, si el clearence es bajo, la vida media del fármaco será mayor. Con estos dos parámetros se pueden diseñar regímenes de dosificación que son útiles para fármacos como antiarrítmicos, teofilina y anticonvulsivantes, es decir, fármacos de rango terapéutico estrecho. Por ejemplo, en el caso de una arritmia, para obtener un efecto inmediato se entrega una dosis de carga y en forma posterior, una dosis de mantención.
La vida media del paracetamol fluctúa entre 4 a 6 horas, por lo que para mantener la concentración plasmática se debe dosificar en intervalos adecuados y administrarlo por lo menos cada vida media y no sólo cuando existe dolor. Cuando no hay buena respuesta terapéutica a los analgésicos, puede ser porque se está administrando a intervalos inadecuados. Entonces, conocer la vida media de un fármaco es útil para diseñar, planificar y racionalizar los regímenes terapéuticos.
Conclusiones respecto a la vida media de un fármaco:
Si un paciente con insuficiencia cardiaca necesita tomar digital, para alcanzar el estado estacionario tendría que recibir una dosis diaria durante 7 días; entonces, se debe entregar una dosis de carga para alcanzar el estado estacionario en forma rápida. En cambio, si se administra digoxina cada vida media, en 7 días se alcanza el estado estacionario, donde la cantidad que ingresa del fármaco es igual a la que egresa.
La vida media de eliminación depende del volumen de distribución y del clearence. Si hay un volumen de distribución alto significa que hay mucho fármaco en los tejidos, o sea, poco fármaco disponible para ser eliminado y así la disminución de la concentración plasmática será gradual.; por lo tanto, si el volumen de distribución es alto, la vida media del fármaco será mayor. A mayorclearence, la vida media del fármaco será menor, porque por cada volumen que el órgano procese se eliminará todo el fármaco contenido en ese volumen. Por el contrario, si el clearence es bajo, la vida media del fármaco será mayor. Con estos dos parámetros se pueden diseñar regímenes de dosificación que son útiles para fármacos como antiarrítmicos, teofilina y anticonvulsivantes, es decir, fármacos de rango terapéutico estrecho. Por ejemplo, en el caso de una arritmia, para obtener un efecto inmediato se entrega una dosis de carga y en forma posterior, una dosis de mantención.
La vida media del paracetamol fluctúa entre 4 a 6 horas, por lo que para mantener la concentración plasmática se debe dosificar en intervalos adecuados y administrarlo por lo menos cada vida media y no sólo cuando existe dolor. Cuando no hay buena respuesta terapéutica a los analgésicos, puede ser porque se está administrando a intervalos inadecuados. Entonces, conocer la vida media de un fármaco es útil para diseñar, planificar y racionalizar los regímenes terapéuticos.
Conclusiones respecto a la vida media de un fármaco:
- Después de cada dosis aumenta la cantidad de fármaco en el organismo, aumenta la cantidad de fármaco eliminado y disminuye la velocidad de acumulación.
- Si el volumen de distribución es grande, se requiere mayor tiempo para alcanzar el estado estacionario y la excreción es lenta.
- Si aumenta el clearence, el tiempo para alcanzar el estado estacionario es menor.
- Si el intervalo de administración del fármaco es menor que la vida media, la acumulación es tóxica.
Estado estacionario
Cuando la cantidad de fármaco que entra al organismo es igual a la que sale, se dice que la concentración plasmática ha alcanzado el estado estacionario.
Un fármaco se acumula en el organismo cuando: la cantidad que ingresa es mayor a la que sale; la velocidad de ingreso es mayor que la velocidad de salida; o se administra una misma dosis a intervalos inferiores a la vida media. Por ejemplo, si a un paciente se le administran 500 mg de paracetamol cada 4 horas, tendrá un estado estacionario; pero si no tiene buena respuesta terapéutica al fármaco y se aumenta la dosis a 1000 mg cada 4 horas, el estado estacionario de esta nueva dosis de paracetamol se logrará dentro de 4 vidas medias; esto ocurre cuando se administra mayor dosis en el mismo intervalo de tiempo, pero si ese paracetamol se administra cada 2 horas, aun cuando sean 500 mg, a las 6 horas el paciente estará intoxicado con el fármaco.
Se puede modificar el intervalo o la dosis, pero existen fluctuaciones entre la dosis máxima y la dosis mínima. Al administrar una dosis constante de un fármaco, se ajusta la dosis a la concentración plasmática media terapéutica, como es el caso de la infusión continua, en que la dosis está en el promedio entre la dosis máxima y la dosis mínima terapéutica; pero si el fármaco se administra por vía oral cada vida media habrá una fluctuación de la dosis, de tal modo que la concentración disminuirá a la mitad y luego se recuperará dicha pérdida, hasta que se acumule. Si las fluctuaciones de la dosis están dentro del rango terapéutico, no habrá problema. A veces se pueden modificar las dosis, aumentándolas y aumentando también los intervalos de administración; esto se puede lograr conociendo los parámetros farmacocinéticos del fármaco, es decir, el volumen de distribución, la vida media, el clearance, entre otros (Fig. 6).
Un fármaco se acumula en el organismo cuando: la cantidad que ingresa es mayor a la que sale; la velocidad de ingreso es mayor que la velocidad de salida; o se administra una misma dosis a intervalos inferiores a la vida media. Por ejemplo, si a un paciente se le administran 500 mg de paracetamol cada 4 horas, tendrá un estado estacionario; pero si no tiene buena respuesta terapéutica al fármaco y se aumenta la dosis a 1000 mg cada 4 horas, el estado estacionario de esta nueva dosis de paracetamol se logrará dentro de 4 vidas medias; esto ocurre cuando se administra mayor dosis en el mismo intervalo de tiempo, pero si ese paracetamol se administra cada 2 horas, aun cuando sean 500 mg, a las 6 horas el paciente estará intoxicado con el fármaco.
Se puede modificar el intervalo o la dosis, pero existen fluctuaciones entre la dosis máxima y la dosis mínima. Al administrar una dosis constante de un fármaco, se ajusta la dosis a la concentración plasmática media terapéutica, como es el caso de la infusión continua, en que la dosis está en el promedio entre la dosis máxima y la dosis mínima terapéutica; pero si el fármaco se administra por vía oral cada vida media habrá una fluctuación de la dosis, de tal modo que la concentración disminuirá a la mitad y luego se recuperará dicha pérdida, hasta que se acumule. Si las fluctuaciones de la dosis están dentro del rango terapéutico, no habrá problema. A veces se pueden modificar las dosis, aumentándolas y aumentando también los intervalos de administración; esto se puede lograr conociendo los parámetros farmacocinéticos del fármaco, es decir, el volumen de distribución, la vida media, el clearance, entre otros (Fig. 6).
Figura 6. Fluctuación de las concentraciones plasmáticas respecto del intervalo y dosis de administración.
Los objetivos de los regímenes de dosificación son: mantener concentraciones terapéuticas de un fármaco en el estado estacionario; individualización de la terapia; mantener la concentración plasmática dentro de rangos apropiados; se utiliza en forma preferente para fármacos con margen terapéutico estrecho como los anticonvulsivantes, antiarrítmicos, digoxina y teofilina. La teofilina y la carbamazepina son capaces de inducir su propio metabolismo mediante la inducción enzimática del citocromo P450, por lo cual se debe establecer un régimen de dosificación considerando este hecho y aumentar las dosis del fármaco cuando se requiera.
La dosis de mantención es la cantidad de fármaco más apropiada para su administración, en infusión continua o en forma intermitente, con el objetivo de mantener el estado estacionario. El valor calculado depende del clearence del fármaco; cuando la administración es vía oral, la dosis dependerá de la vida media del fármaco y se obtiene del producto del clearence por la concentración plasmática. Si se multiplica la dosis de mantención por la vida media, el resultado es el intervalo de administración del fármaco.
La dosis de carga es la cantidad de fármaco necesaria para alcanzar en forma rápida la concentración deseada, dentro del rango terapéutico. Significa que se llenarán los depósitos físicos con el fármaco en forma aguda. Siempre existe el riesgo de alcanzar concentraciones plasmáticas tóxicas de una sola vez, por esta razón la digoxina se administra fraccionada y no en bolo. La dosis de carga depende del volumen de distribución del fármaco.
La dosis de mantención es la cantidad de fármaco más apropiada para su administración, en infusión continua o en forma intermitente, con el objetivo de mantener el estado estacionario. El valor calculado depende del clearence del fármaco; cuando la administración es vía oral, la dosis dependerá de la vida media del fármaco y se obtiene del producto del clearence por la concentración plasmática. Si se multiplica la dosis de mantención por la vida media, el resultado es el intervalo de administración del fármaco.
La dosis de carga es la cantidad de fármaco necesaria para alcanzar en forma rápida la concentración deseada, dentro del rango terapéutico. Significa que se llenarán los depósitos físicos con el fármaco en forma aguda. Siempre existe el riesgo de alcanzar concentraciones plasmáticas tóxicas de una sola vez, por esta razón la digoxina se administra fraccionada y no en bolo. La dosis de carga depende del volumen de distribución del fármaco.
No hay comentarios:
Publicar un comentario