La alcaptonuria (enfermedad de la orina negra u oscura) es una enfermedad hereditaria rara caracterizada por un trastorno del metabolismo de la tirosina y la fenilalanina. Se hereda con un patrón autosómico recesivo y causada por un defecto en el gen HGD que codifica a la enzima homogentisato-1,2-dioxigenasa (EC 1.13.11.5).1 El resultado es la acumulación de uno de los productos tóxicos de la ruta metabólica en cuestión, una molécula llamada ácido homogentísico, el cual circula por la sangre y se excreta en laorina en grandes cantidades, dándole la característica coloración negruzca a la orina. El exceso del ácido homogentísico causa daño a los cartílagos, una condición llamada ocronosis, que conlleva a osteoartritis, a lasválvulas del corazón y produce cálculos renales. La alcaptonuria es frecuente en Eslovaquia y en la República Dominicana, con mayor incidencia que otros países.
Diagnóstico
El diagnóstico más frecuente se basa en la detección de niveles significativos de ácido homogentísico (HGA) en la orina, mediante análisis cromatográfico. Otra forma de diagnósticar la enfermedad es mediante la secuenciación del gen HGD, para observar si existe alguna mutación en él.
Tratamiento
Es necesario tratar el dolor articular que sufren las personas con alcaptonuria. También es importante una terapia física, para fortalecer la musculatura y la flexibilidad. Si el dolor articular es muy severo, es posible realizar cirugía de reemplazo para aliviarlo. Es recomendable seguir una dieta pobre en fenilalanina y tirosina, pero rica enácido ascórbico.
Está en estudio el tratamiento por administración de nitisinona, el cual suprime la producción del ácido homogentísico.
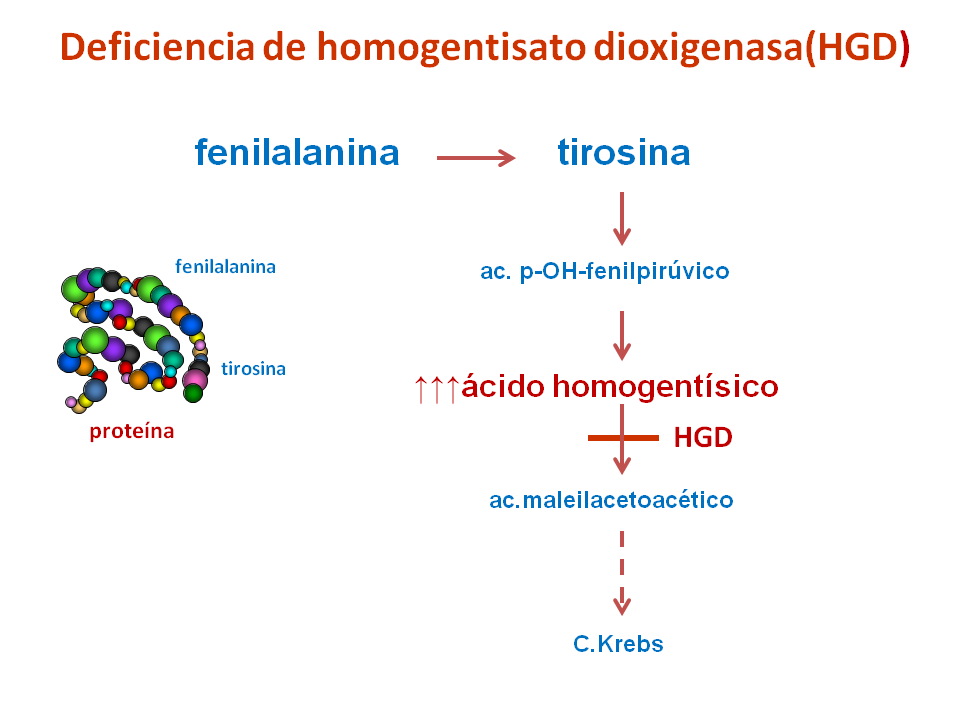
La alcaptonuria es un error congénito del metabolismo de los aminoácidos fenilalanina y tirosina, causado por la deficiencia de la enzimahomogentisato dioxigenasa (HGD), que determina la acumulación de ácido homogentísico en sangre y orina.
Se caracteriza por la orina de color oscuro, ocronosis (pigmentación del tejido conjuntivo) y artrosis degenerativa de las articulaciones.
¿Qué es el ácido homogentísico?
Es un compuesto intermedio de la vía de degradación de unos aminoácidos (fenilalanina y tirosina) hacia el ciclo de Krebs. Es el sustrato de la enzima HGD y, en condiciones normales, es prácticamente indetectable en sangre y orina.
¿Cuándo se acumula el ácido homogentísico?
Cuando dicha vía catabólica (de degradación) está interferida por una deficiencia enzimática de HGD, el ácido homogentísico se acumula en sangre y se elimina en grandes cantidades en la orina.
En contacto con el aire el ácido homogentísico se oxida y polimeriza, dando lugar al pigmento negro alcaptón, que da color a la orina de los individuos que padecen esta enfermedad, a la que da el nombre (alcapton-uria).
El pigmento se deposita también en tejido conjuntivo (ocronosis), dándole un aspecto grisáceo y causando su degeneración.
Cuando se deposita en las articulaciones causa una artropatía degenerativa dolorosa y discapacitante.
¿Por qué se produce un defecto de la enzima HGD?
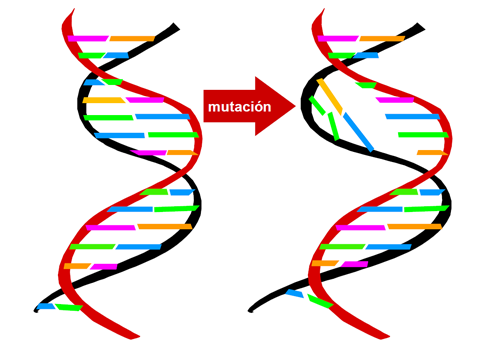
La deficiencia de HGD se produce debido a mutaciones (cambios estables y hereditarios) en el gen que codifica esta proteína enzimática, el gen HGO.
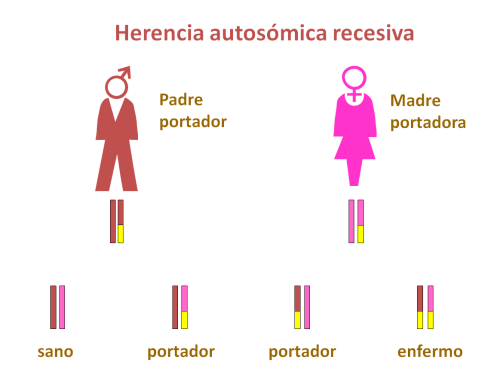
La alcaptonuria se transmite de forma autosómica recesiva, es decir, ambos padres son portadores de una mutación en el gen HGO, aunque no padecen ninguna manifestación clínica por ello.
Si ambos padres pasan al hijo un alelo mutado de este gen, el niño sufrirá una alcaptonuria.
La enfermedad de Alexander es una enfermedad genética extremadamente rara, normalmente de aparición en la infancia y perteneciente al grupo de las leucodistrofias. Este grupo de enfermedades neurológicas se caracteriza por la destrucción progresiva de la sustancia blanca del cerebro. La enfermedad de Alexander se manifiesta por la aparición de retraso mental y anormalidades físicas, en especial macrocefalia, por la presencia de fibras de Rosenthal y patrones de neuroimagen característicos. La enfermedad progresa hasta un desenlace mortal en la mayor parte de los casos. La enfermedad recibe el nombre por el patólogo neozelandés William Stuart Alexander, quien describió el síndrome en 1949 junto con la profesora Dorothy Rusell en el London Hospital.
| Enfermedad de Alexander | ||
|---|---|---|
 Imagen de una leucodistrofia en una niña de 18 meses. Posible Enfermedad de Alexander |
Epidemiología
La prevalencia de la enfermedad es desconocida. La mayor parte de los casos se dan de forma esporádica, sin que exista un historial familiar de propensión a la enfermedad. Entre otras cosas, esto significa que los padres que tienen un hijo con la enfermedad, tienen una probabilidad muy baja de que siguientes hijos la tengan, sin embargo, existen algunas familias con más de un hijo afectado. Es posible que exista una heredabilidad de la propensión a padecer mutaciones "de novo" en general en la descendencia. Así mismo, existen dos casos de familias en que la enfermedad se hereda de forma dominante.2 En ambos, la aparición es posterior a los 25 años. Hasta 2005 existían tan solo 57 casos descritos. Se han publicado 500 casos desde su descripción en 1949.3 La enfermedad es más frecuente en mujeres (proporción 3:1).es una enfermedad degenerativa
Etiología

En principio, se observó la acumulación en el cerebro en torno a los astrocitos y a la barrera hematoencefálica de unas estructuras características eosinófilas y vermiformes que se llamaron fibras de Rosenthal, en honor al patólogo alemán Werner Rosenthal quien las describió en 1989 en lasiringomielia. Estas estructuras eran ricas en PAFG y en una proteína de shock término llamada α-B-Cristalina.
La alteración subyacente fue caracterizada en 2001 por Messing y colaboradores.4 Se trata de una enfermedad de carácter genético autosómica dominante, originada en la mayor parte de los casos por una mutación puntual de novoen el gen de la proteína ácida fibrilar glial. Este gen está situado en la banda q21 del cromosoma 17 (17 q21). Li y colaboradores observaron que en 24 de 28 casos de su estudio, el cromosoma portador es el paterno, con lo que parece que la mutación se da mucho más en la espermiogénesis que durante el desarrollo embrionario. Parece ser que no hay una relación con la edad del padre.5 Se han descrito más de 49 mutaciones o alelos patológicos que pueden desencadenar la enfermedad:
La alteración subyacente fue caracterizada en 2001 por Messing y colaboradores.4 Se trata de una enfermedad de carácter genético autosómica dominante, originada en la mayor parte de los casos por una mutación puntual de novoen el gen de la proteína ácida fibrilar glial. Este gen está situado en la banda q21 del cromosoma 17 (17 q21). Li y colaboradores observaron que en 24 de 28 casos de su estudio, el cromosoma portador es el paterno, con lo que parece que la mutación se da mucho más en la espermiogénesis que durante el desarrollo embrionario. Parece ser que no hay una relación con la edad del padre.5 Se han descrito más de 49 mutaciones o alelos patológicos que pueden desencadenar la enfermedad:
- Diana Rodríguez y colaboradores encontraron en nueve pacientes mutaciones en el dominio central en forma de bastón de la proteína, habitualmente en los segmentos 1A, 2A y 2B. La mayor parte daban cambios en residuosarginina (cuatro en la posición R79H, cuatro en R239C y una en R239H, 2R88C y 1R88S, estas dos últimas desconocidas hasta ese momento). Los dos casos restantes del estudio afectaban a otros aminoácidos (1L76F y 1N77Y). El aminoácido afectado parecía determinar la severidad del curso de la enfermedad.6
- En 2007 Meins y colaboradores detectaron otras mutaciones puntuales en otro dominio (cuya posición está próxima al C terminal de la proteína) que determinaban la aparición temprana de la enfermedad, en concreto A364 V y Y366C. Se observó que mutaciones idénticas en otros filamentos intermedios, por ejemplo en la queratina, daban lugar a problemas similares, con lo que se sugiere que estas secuencias son críticas para la estabilidad de estos filamentos.7
Patogenia
Hasta la fecha, el mecanismo propuesto más aceptado para explicar la enfermedad sería el siguiente:8
- La acumulación de proteína ácida fibrilar glial (PAFG) y la consiguiente formación de agregados característicos, denominados fibras de Rosenthal en varios tipos celulares, y en especial los astrocitos. Parece que la acumulación se debe a una ganancia de función por causa de la mutación que bloquea parcialmente el ensamblaje de los filamentos de PAFG.9
- Secuestro posterior de ubiquitina y las proteínas chaperonas α-B-cristalina y HSP27 en las fibras de Rosenthal.
- Activación tanto de la proteína Jnk como de la respuesta de estrés.
Anatomía patológica

La enfermedad de Alexander es primariamente una alteración de los astrocitos, que forman parte de las células de soporte de las neuronas (neuroglía). Entre los astrocitos, los más comunes en la sustancia blanca son los de tipo fibroso.10 Estos últimos se caracterizan porque en su citoesqueleto contienen un filamento intermedio, la ya mencionada PAFG, que al mutar construye una estructura proteica defectuosa. Esta se almacena junto con la ubiquitina y otras dos proteínas de shock térmico, las ya conocidas fibras de Rosenthal. Se pueden localizar en todo el sistema central, tanto en el cerebro como en la médula espinal, pero en especial en la vecindad de los vasos sanguíneos de la superficie del cerebro. Las imágenes por microscopía electrónica muestran un vínculo estrecho entre las fibras de Rosenthal y los filamentos intermedios. Además existe desmielinización histológicamente hablando en los afectados tardíos, o ausencia de mielinización en los niños. Se encuentran afectadas igualmente las fibras sensoriales y las motoras. En la aparición temprana con macrocefalia es común la degeneración de la sustancia blanca y a veces se acompaña también de hidrocefalia.
Sin embargo, las áreas desmielinizadas no coinciden con la distribución de las fibras de Rosenthal, por ello parece que la desmielinización y la aparición de fibras parecen ser manifestaciones independientes de la enfermedad. No obstante se asume que la desmielinización se produce por la degeneración final de los astrocitos.
| Tabla1: CUADRO CLÍNICO de enfermos diagnosticados mediante MRI de enfermedad de Alexander en una muestra de 5 y 14 pacientes respectivamente, según van der Knaap y otros11 | ||
|---|---|---|
| Signos observados | Variante infantil | variante juvenil |
| Nº de pacientes | 5 | 14 |
| Edad de primeras manifestaciones | nacimiento a 4 semanas | <2 a="" os="" th=""> |
| Retraso desarrollo motor | severo(5) | severo(2)moderado(11)no observado(1) |
| Principal indicador motor | ninguno(3)asir(2) | marcha(12)sentarse(2) |
| Retraso desarrollo mental | Severo(5) | moderado(13) |
| Problemas de conducta | 2 | 9 |
| Epilepsia | Severa (5) | Moderada (11) |
| Probemas de alimentación | 5 | 12 |
| Vómitos | 5 | 6 |
| Dificultades para tragar/asfixia | 4 | 11 |
| ganancia peso insuf | 5 | 5 |
| Problemas de habla | 2, no aplicable en 3 | 14 |
| Macrocefalia | 4,98º percentil en 1 | 9,98º percentil en 2/no obs. en 3 |
| Pobre contacto visual | 5 | 0 |
| Hipotonía | Generalizada (2) axial(3) | Generalizada (3) |
| Hipertonía de miembros | 3 | 8 |
| Hiperreflexia | 5 | 14 |
| Ataxiacerebelar | No comprobable | 12 |
| Signosextrapiramidales | 3 | 3 |
| Deterioro | rápido(3)moderado(2) | lento(11)moderado(1) |
| Edad de fallecimiento | 4 meses a 2¹/2 años | 7-18 años en 4 casos |
Cuadro clínico
La enfermedad tiene varias formas que difieren en su cuadro clínico y en la edad de aparición, teniendo todas ellas el rasgo distintivo de la degeneración fibrinoide de los astrocitos con fibras de Rosenthal. Desde 1976 se reconocen tres formas de la enfermedad:12
- Forma infantil:
-
- Aparece desde el nacimiento hasta los 2 años.
- Es la más común.
- Puede cursar con o sin macroencefalia, aunque lo más corriente es que aparezca (Rodríguez, 2001).
- Se observan ataques y retraso o involución en el desarrollo. La función motora se deteriora progresivamente hasta la cuadriparesis y espasticidad.
- Hidrocefalia, en ocasiones asociada a estenosis del acueducto de Silvio.13 Aunque no se ha observado asociación entre la estenosis y la acumulación de fibras de Rosenthal.
- Retraso mental profundo en la mayoría de los casos, aunque a veces no se ha observado.
- Ataxia y ataques epilépticos.
- vómitos y elevada tensión intracraneal.
- Forma juvenil:14
-
- Comienza en la edad escolar, a los 9,5 años de media
- La sintomatología consiste principalmente en paraplejia espástica y signos bulbares progresivos.
- Normalmente se preservan las funciones cognitivas.
- Dificultades para tragar o hablar, vómitos, ataxia y/o espasticidad.
- Puede darse cifoscoliosis
- Mientras la forma infantil afecta generalmente al cerebro, la juvenil afecta más al tronco cerebral. Hay muchas fibras de Rosenthal, pero la desmielinización es menos importante que en la forma infantil.
- Forma adulta:
-
- Es la forma más benigna y rara de la enfermedad. Se han observado casos de aparición hasta los 36 años. Recuerda a la esclerosis múltiple o un tumor.
- La ataxia es muy frecuente, así como dificultades de habla, deglución y problemas en el sueño.
- Los síntomas son similares a la enfermedad juvenil, pero más suaves.
Diagnóstico
El diagnóstico había sido durante mucho tiempo difícil, debido a que la mayoría de los signos podían darse también en otras leucodistrofias, de modo que la prueba diagnóstica de confirmación consistía en una biopsia cerebral que revelara las fibras de Rosenthal. En ocasiones esto se efectuaba post mórtem en la necropsia. Sin embargo, esta prueba resulta bastante invasiva y se vio la necesidad de buscar otras posibilidades aprovechando las nuevas técnicas de imagen por MRI. En marzo de 2001 la eminente neuróloga Marjo van der Knaap y su equipo pudieron establecer una serie de criterios que probaban en un 90% de los casos la existencia de la enfermedad.11 Se considera que la presencia de al menos 4 de los siguientes criterios dan un resultado diagnóstico positivo:
- Presencia de anormalidades extensas en la materia blanca con una preponderancia frontal o bien en cuanto a la extensión de estas anormalidades, en el grado de abultamiento, en el grado de cambio de señal o en el de pérdida de tejido (por atrofia o degeneración cística).
- Presencia de un borde periventricular de descenso de intensidad de señal en las imágenes potenciadas en T2 y elevación de la intensidad en imágenes potenciadas en T1.
- Anormalidades en los ganglios basales y tálamos, consistentes en una elevación en la intensidad de la señal y abultamiento o en atrofia y aumento o descenso de la intensidad de la señal en imágenes potenciadas en T2.
- Anormalidades en el tronco encefálico, en particular incluyendo el cerebro medio y la médula.
- Aumento de contraste que implica uno o más de las siguientes estructuras: borde ventricular, cerco
- de tejido periventricular, sustancia blanca de los lóbulos frontales, quiasma óptico, fórnix, ganglis basales, tálamo, núcleo dentado y estructuras troncoencefálicas.
La enfermedad de Alexander
La enfermedad de Alexander fue identificada en 1949 en los criterios neuropatológicos, es decir, la presencia de fibras de Rosenthal y la pérdida de la mielina.
Desde entonces, diferentes formas clínicas han sido individualizadas.
La forma infantil (desde el nacimiento hasta los 2 años) es la más común, se caracteriza por un inicio temprano y su evolución severa. Sus síntomas asociados son macrocefalia progresiva (a veces hidrocefalia), retraso del desarrollo psicomotor o deterioro mental, signos piramidales, ataxia y convulsiones. El escáner y la RMI en el diagnóstico presentan anomalías en la sustancia blanca, predominantemente en las regiones frontales.
La forma juvenil comienza en la edad escolar con paraplejia espástica progresiva y signos bulbares asociados.
Los casos de adultos son heterogéneos y difíciles de diagnosticar.
Esta rara enfermedad, a menudo considerada como una leucodistrofia, es normalmente esporádica y sólo unos pocos casos familiares han sido localizados.
El descubrimiento de las fibras de Rosenthal en ratones transgénicos que sobreexpresan GFAP (proteína glial fibrilar ácida) en humanos, ha impulsado la búsqueda de mutaciones.
Más de 20 mutaciones han sido identificadas en la secuencia de codificación de la GFAP. Estas son mutaciones de novo dominantes.
Sin embargo, el diagnóstico prenatal parece conveniente dado el riesgo de mosaicismo germinal.
En la actualidad, el tratamiento es sólo sintomático.
No hay comentarios:
Publicar un comentario