Anomalías estructurales cromosómicas
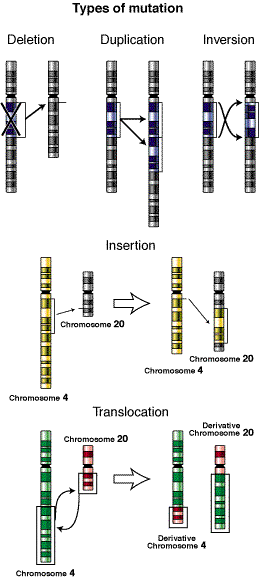
duplicación cromosómica es la repetición de un fragmento de cromosoma a continuación del fragmento original. Las duplicaciones surgen por error en laduplicación del ADN, como producto de una reorganización cromosómica de tipo estructural o relacionado con un proceso de sobrecruzamiento defectuoso. Las duplicaciones no suelen ser deletéreas, son una fuente de nuevo material genético y base para nuevos cambios evolutivos. Muchas de las familias génicas con un origen evolutivo común, o las familias multigénicas pueden tener su origen en las duplicaciones. Si el segmento afectado es de gran tamaño, se puede detectar en meiosis con los mismos criterios que en las deleciones(bivalente heteromorfo o zona intersticial desapareada en el cromosoma con la duplicación).
Las duplicaciones no suelen tener una manifestación fenotípica observable a simple vista, sino mediante análisis citogenéticos y moleculares.
Papel de las duplicaciones en la evolución
Uno de los aspectos integrantes del estudio de la evolución es especular sobre los mecanismos posibles de la variación genética. En 1970, Susumo Ohno publico el polémico libro Evolution by Gene Duplication. La tesis de Ohno se basaba en la suposición de que los productos de genes esenciales son indispensables para la supervivencia de los miembros de cualquier especie a lo largo de la evolución. Estos genes no pueden acumular mutaciones que alteren su función primaria y dar lugar potencialmente a nuevos genes.1 Sin embargo, si se duplicara un gen esencial en una línea germina, en la copia extra se tolerarían cambios mutacionales proporciona la información genética para su función esencial. La copia duplicada quedaría libre para adquirir muchos cambios mutacionales durante largos periodos de tiempo. En periodos cortos, la nueva información genética podría no tener ventajas prácticas. Sin embargo, en periodos evolutivos largos, el gen duplicado podría cambiar lo suficiente como para que su producto asumiera un papel divergente en la célula. La nueva función podría dar una ventaja “adaptativa” al organismo, incrementando su eficacia biología. Ohno ha imaginado un mecanismo mediante el cual pudo haberse originado sustancial variabilidad genética. La tesis de Ohno está apoyada por el descubrimiento de genes que tiene una parte importante de sus secuencias de ADN en común, pero cuyos productos génicos son distintos.
La importancia evolutiva de las duplicaciones radica en el hecho de que los individuos portadores tienen dos copias de un mismo gen. En un individuo normal una mutación de ese gen puede tener efectos deletéreos, pero si hay dos copias y se produce una mutación en una de ellas, el individuos podrá seguir manifestando un fenotipo "aparentemente normal" y la selección natural no actuaría en su contra. Mediante este proceso se pueden ir originando nuevas copias de un mismo gen y producirse variantes y alternativas no alélicas a una secuencia de ADN. Este es el origen de las familias multigénicas (Histonas, rRNAs, etc.) y de las familias génicas con un origen evolutivo común (Ej, haptoglobinas). La estructura citogenética de las familias multigénicas suele ser muy típica: Todos los genes que componen la familia se encuentran juntos en el cromosoma en un mismo "nicho" o cluster, que a su vez puede estar repetido una o varias veces.
Introducción técnica
Síndrome de Inversión-Duplicación del cromosoma 15q.
Consulte el artículo original aquí
Introducción
El Síndrome de inversión-duplicación del cromosoma 15q, es un síndrome identificable clínicamente, ocasionado por duplicaciones de la región q11-q13 del cromosoma 15. Está relacionado con trastornos de espectro autista, retraso madurativo, dificultades en el aprendizaje, trastornos del desarrollo y convulsiones-epilepsia.
Las manifestaciones iniciales de este síndrome, son hipotonía central temprana, pequeñas características físicas, trastorno significativo del desarrollo, comportamientos autísticos, convulsiones infantiles o epilepsias de difícil control.(1)
Las duplicaciones-inversiones en el cromosoma 15q generalmente suceden de una de las dos formas siguientes: Como un cromosoma isodicéntrico extra o como una duplicación intersticial del cromosoma 15q.
Cromosoma 15q isodicéntrico
El cromosoma 15 Isodicéntrico, abreviado idic(15), se diagnostica en personas que tienen 47 cromosomas (o en algunos casos, más) en lugar de los habituales 46. El cromosoma extra se genera a partir de una porción del cromosoma 15 que se ha duplicado e invertido. De esa forma hay dos copias parciales del cromosoma 15q, unidas entre sí, que parecen ser imágenes especulares. A causa de esta configuración, Idic15 habitualmente se refiere a una "inversión-duplicación del 15q". Por lo general, es la región llamada 15q11-q13 la que se duplica. En algunos casos la región afectada es aún mayor. El tamaño del idic(15) depende del tamaño de la región del cromosoma 15 que se duplica (2).
Duplicación intersticial del 15q
Personas nacidas con los 46 cromosomas habituales, pero que tienen un segmento de material duplicado en el interior del cromosoma 15. Se abrevia como invdup15. Con frecuencia la sección duplicada (15q11-q13) es la misma que la de los casos con idic15. Por este motivo las personas con idic15e invdup15 comparten características. Dentro del grupo, las personas con invdup15 manifiestan síntomas más leves que aquellas con idic15.
Cuando el material genético extra procede del cromosoma paterno, un niño puede seguir un desarrollo típico. En aquellos casos en los que el material duplicado procede del cromosoma materno, generalmente surgirán los problemas de desarrollo. En la mayoría de los casos, este síndrome no es hereditario, se produce en algún momento durante la formación de las células reproductivas (Esperma y óvulo).
Diagnóstico
Se confirma mediante técnicas citogenéticas standar y análisis FISH, que confirmarán el diagnóstico distinguiendo el dup15q de marcadores de cromosomas supernumerarios. Las duplicaciones intersticiales son más complicadas de detectar con análisis rutinarios pero son claramente visibles usando un estudio FISH 15q o un array/microarray clínico CGH. Los estudios a nivel molecular como el análisis de ADN microsatélite en los padres o estudios de metilación del ADN del niño afectado, determinarán la procedencia paterna o materna del dup15q(q). Las familias siembre deben interpretar los resultados de las pruebas genéticas con médicos y/o especialistas en genética para asegurar una interpretación adecuada de los resultados.
Clínica
Aunque actualmente existen unos 100 casos y estudios disponibles en la literatura científica, la información disponible acerca del síndrome de inversión/duplicación del cromosoma 15 es muy limitada. Especialmente en adultos
Existe un rango muy amplio en el grado de afectación y en los problemas de desarrollo experimentados en personas con el síndrome idic15. Dos personas con el mismo patrón cromosómico pueden ser manifestar capacidades muy diferentes. La literatura científica no ha encontrado relación clara entre el tamaño de la región duplicada y la severidad de los síntomas. Describiremos en las secciones siguientes información sobre características físicas, trastornos en el desarrollo, cuestiones médicas y tratamientos para el síndrome de inversión-duplicación del cromosoma 15.
Características físicas.
A diferencias de la mayoría de otros síndromes cromosómicos, existen pocas características físicas reseñables que pueden atribuirse al síndrome de duplicación del cromosoma 15q. Los hallazgos físicos son muy inespecíficos y pueden incluir los siguientes:
- Hipotonía: Los bebés con Idic15 frecuentemente tienen hipotonía (tono muscular disminuido). Se muestran laxos y pueden mostrar dificultades a la hora de succionar y alimentarse. Los hitos del desarrollo motriz, como los volteos, sedestación, bipedestación y la marcha se retrasan significativamente. Los niños mayores y los adultos con hipotonía se cansan con facilidad. La hipotonía en idic15 frecuentemente, remite con la edad y evoluciona a hipertonía (tono muscular aumentado). Particularmente en la parte inferior de las piernas.
- Manifestaciones físicas: Muchos de los niños/as con idic15 comparten características faciales. La nariz suele achatarse, en la zona del puente nasal que les genera una nariz “botón”. Pueden manifestarse pliegues en la piel junto a la zona interna de los ojos, llamadas “epicanto”. Igualmente los ojos pueden estar un poco hundidos. Las orejas pueden ser pequeñas y rotadas hacia la parte posterior y pueden existir pliegues notables el los bordes de las mismas. El paladar (ojival) puede ser inusualmente alto. El labio superior está con frecuencia algo levantado dejando visible parte de la dentadura superior. También hay algunos datos acerca de zonas de incremento o decremento de la pigmentación de la piel.
- Crecimiento: El crecimiento está en torno a un 20-0% por debajo de las personas con desarrollo típico, sin embargo el perímetro craneal suele permanecer dentro de los límites normales.
- Otras características: Con menor frecuencia, los bebés con idic15 pueden nacer con fisuras en labios o en el paladar (labio leporino) además de diferencias en el modo en que se desarrolla su corazón (defectos en tabicación), riñones (agenesias, digenesias) u otros órganos durante la embriogenesis. Por este motivo es importante que los niños con idic15 de reciente diagnóstico sean evaluados detalladamente a causa de estas posibles diferencias estructurales. El Hipogonadismo, incluyendo el no descenso testicular(cliptorquídia) se manifiesta aproximadamente en un 20% de los casos afectados. Aunque la pubertad generalmente se da con normalidad en la mayoría de los casos, algunos desórdenes como un desarrollo demasiado temprano se ha observado con frecuencia en el género femenino. Se recomienda un examen completo del aparato genitourinario en niños diagnosticados con idic15.
Dificultades en el desarrollo en idic(15)
- Retrasos en la motricidad gruesa. Son muy frecuentes y en parte están relacionados con la hipotonía. En el 2005 un estudio científico puso de manifiesto que la sedestación generalmente se adquiría entre los 10 y los 20 meses, la marcha a pie en torno a los 2-3 años (1). Otro estudio algo más reciente, puso de manifiesto que niños con duplicaciones isodicéntricas consiguen caminar de forma independiente a una edad media de 25.5 meses (13-54 meses, incluyendo tres casos de 47 que no eran “ambulatorios” en el momento de las pruebas (3). La mayoría de las personas con idic15 pueden caminar de forma independiente a pesar de que se evidencie un cierto grado de ataxia (problemas de coordinación).
- Retrasos en la motricidad fina. La información que llega de las familias sugieren que las dificultades en la motricidad fina es muy frecuente en niños y niñas con idic15. El uso no funcional de los objetos unido a un modo inmaduro de explorar se ponen de manifiesto en la literatura científica (1)
- Discapacidad intelectual. La mayoría de los individuos con idic15 muestran algún tipo de trastorno del desarrollo mental y dificultades de aprendizaje. En los casos más afectados se incluye retraso mental.
- Trastornos del Espectro Autista (TEA). Son muchos los informes que reportan el riesgo de TEA en personas con idic15q, si bien, no todos los casos desarrollan autismo. Dos estudios sobre una muestra de 226 pacientes con autismo encontró idic15 aproximadamente en un 3-5% de los casos (4,5). Duplicaciones en el cromosoma 15q11-q13 son la cromosomopatía más frecuente en personas con autismo.
- Problemas en la comunicación y el lenguaje. La mayor parte de los niños con idic15 están afectados por retraso en la comunicación y el lenguaje. El lenguaje expresivo puede ser muy pobre o incluso no darse, y generalmente es ecolálico (ecolalias inmediatas o retardadas) y reversión pronominal(1) en su estudio del idic(15) la Dra. Carolyn Schanen encontró que 26 de 47 niños disponían de algo de lenguaje en el momento de su participación en el estudio, la primera palabra se consiguió a un promedio de 28,7 meses (rango entre 7-84 meses) y la construcción de frases a una media de 44,1 mes (rango entre 9 -114 meses). Mientras la mayoría de los casos tienen estas dificultades un pequeño subconjunto de niños con idic15 son altamente verbales.
- Problemas en el procesamiento sensorial La información procedente de los padres sugiere que los problemas en el procesamiento sensorial son muy frecuentes. Estas dificultades impactan sobre la capacidad de los niños de conseguir un grado de adaptación correcta los desafíos de la vida diaria. Los umbrales de reacción pueden estar tanto aumentados como reducidos.
- Comportamiento Son habituales las dificultades en el comportamiento y la comunicación social. Suelen ser bajas las respuestas ante estímulos sociales. En individuos mayores se sugieren evoluciones positivas en este sentido.
Cuestiones médicas relativas al síndrome de duplicación del idiq(15)
- Convulsiones y epilepsia. La epilepsia representa una importante característica clínica del idic15. Más de la mitad de las personas con idic15sufren al menos de un espasmo o crisis a lo largo de su vida. La mayoría de los episodios ocurren antes de los 5 años, pero pueden prolongarse hasta después de la adolescencia. Existen diferentes tipos de espasmos que puede experimentar una persona con idic15. A lo largo de la vida pueden darse diferentes tipos de crisis/espasmos. La prevalencia de convulsiones infantiles sobre los grupos de estudio suele ser muy alta y sugiere una relación bastante clara.(7). Los espasmos infantiles asociados con las hipsarritmias (desorganizaciones) de los EEG (Electroencefalogramas) han sido ampliamente documentados (8). El Síndrome de Lenox-Gastaut o aproximaciones al mismo han sido observados en los cuatro pacientes con idic15 estudiados (Bataglia et al -9). Sufrían espamos tónicos/atónicos (Caida de la cabeza o desvanecimientos), espasmos tónico-clónicos y ausencias atípicas entre los 4 y los 8 años. Crisis complejas parciales y mioclonías se observan igualmente en otros individuos afectados(10).
La respuesta a los tratamientos es muy dispar. Para algunos niños el control de las crisis llega muy rápidamente con medicación y para otros, a pesar del tratamiento antiepiléptico correcto, el control de complica demasiado. Esta dificultad se asocia con algún grado de deterioro asociado. - Trastornos de déficit de atención. También se ha reportado casos asociados a personas con idic15.
- Ansiedad También han sido reportados por padres. Se requiere una mayor profundización en este aspecto.
- Mayor riesgo de muerte súbita Existe un riesgo mayor de muerte repentina, inesperada y hasta el momento inexplicable en niños/as y jóvenes de 7 años de edad en adelante con síndrome de idic15. El riesgo es pequeño y está estimado en 0.5-1% por persona/año. Los terapeutas deben permanecer alerta ante síntomas potenciales y evaluar a sus pacientes de acuerdo con su mejor juicio clínico. Las Benzodiazepinas y barbitúricos sólo deben usarse si no hay otras alternativas, puesto que existe una posible asociación con la muerte súbita en este trastorno concreto. Para más información consulte “Muerte Súbita y Síndrome de Duplicación 15q11-q13”en esta misma web.
- Otros problemas médicos. Se reportan infecciones respiratorias recurrentes en la infancia, supuración del oído medio que pueden requerir drenajes, eccemas, pubertad precoz y adelanto en el desarrollo y otras irregularidades menstruales, exceso de apetito y peso. También abundan casos de escoliosis en la adolescencia.
Tratamientos
En el momento actual no existen tratamientos específicos que pueden directamente abordar los cambios de origen genético observados en personas conidic15. Aunque el desequilibrio genético no puede ser revertido, existen terapias para ayudar en muchos de los síntomas asociados. Trabajo físico y motór, Terapias musicales junto con una correcta intervención educativa que no debe descuidar la parte física, son críticos para niños con idic15 para desarrollar su máximo potencial.
Recomendaciones médicas
Debemos permanecer alerta ante el hecho de que las personas con idic15 pueden reaccionar a la medicación de forma diferente, y ser más sensibles a los efectos secundarios para algunos principios activos (13). Es por ello que los medicamentos deben ser utilizados con especial cuidado. Cualquier medicamento nuevo debe ser introducido en entornos controlados con rampas de dosificación hasta llegar a la dosis terapéutica y con un objetivo claro acerca de cómo se espera que funcionará el tratamiento. Esto incluye también suplementos nutricionales y homeopáticos.
Un grupo de la Univesidad de California, Davis, he registrado tres familias en las que hay cinco personas con duplicaciones intersticiales del cromosoma 15. Los niños padecían TDAH (Trastorno por déficit de atención e hiperactividad), TGD (Trastornos generalizados del desarrollo) y autismo. 3 de 5 fueron tratados con metilfenidato (rubifen) para los síntomas de TDAH con una respuesta adecuada. Uno de los tres chicos fue tratado experimentalmente con Adderall (compuesto de diferentes anfetminas) pero no respondió adecuadamente, por lo que volvió al metilfenidato. Con la Risperidona (risperdal) los resultados fueron dispares. Para un niño fue beneficiosa y para otro la respuesta fue pobre (sin más detalles). La Fluoxetina (Prozac) no fue beneficiosa para ninguno de los 3 niños tratados con ella. Dos manifestaron conductas agresivas y el otro simplemente no respondió a ella. En otros ensayos clínicos la Fluexitina y otros inhibidores de la recaptación de la serotonina (SSRIs) han sido útiles para el tratamiento de la irritabilidad en comportamientos compulsivos, sin embargo en el caso del idic15 debemos tener en mente que muchos de los casos sufren trastornos del comportamiento que deben ser tratados con estabilizadores del mismo antes de usar SSRI. Aunque aún hay mucho trabajo por hacer y queda mucho por investigar, la Oxcarbazepina parece ser más útil para la estabilización del comportamiento que el ácido valproico (Depakine).
En el momento actual no existen tratamientos específicos que pueden directamente abordar los cambios de origen genético observados en personas conidic15. Aunque el desequilibrio genético no puede ser revertido, existen terapias para ayudar en muchos de los síntomas asociados. Trabajo físico y motór, Terapias musicales junto con una correcta intervención educativa que no debe descuidar la parte física, son críticos para niños con idic15 para desarrollar su máximo potencial.
Recomendaciones médicas
Debemos permanecer alerta ante el hecho de que las personas con idic15 pueden reaccionar a la medicación de forma diferente, y ser más sensibles a los efectos secundarios para algunos principios activos (13). Es por ello que los medicamentos deben ser utilizados con especial cuidado. Cualquier medicamento nuevo debe ser introducido en entornos controlados con rampas de dosificación hasta llegar a la dosis terapéutica y con un objetivo claro acerca de cómo se espera que funcionará el tratamiento. Esto incluye también suplementos nutricionales y homeopáticos.
Un grupo de la Univesidad de California, Davis, he registrado tres familias en las que hay cinco personas con duplicaciones intersticiales del cromosoma 15. Los niños padecían TDAH (Trastorno por déficit de atención e hiperactividad), TGD (Trastornos generalizados del desarrollo) y autismo. 3 de 5 fueron tratados con metilfenidato (rubifen) para los síntomas de TDAH con una respuesta adecuada. Uno de los tres chicos fue tratado experimentalmente con Adderall (compuesto de diferentes anfetminas) pero no respondió adecuadamente, por lo que volvió al metilfenidato. Con la Risperidona (risperdal) los resultados fueron dispares. Para un niño fue beneficiosa y para otro la respuesta fue pobre (sin más detalles). La Fluoxetina (Prozac) no fue beneficiosa para ninguno de los 3 niños tratados con ella. Dos manifestaron conductas agresivas y el otro simplemente no respondió a ella. En otros ensayos clínicos la Fluexitina y otros inhibidores de la recaptación de la serotonina (SSRIs) han sido útiles para el tratamiento de la irritabilidad en comportamientos compulsivos, sin embargo en el caso del idic15 debemos tener en mente que muchos de los casos sufren trastornos del comportamiento que deben ser tratados con estabilizadores del mismo antes de usar SSRI. Aunque aún hay mucho trabajo por hacer y queda mucho por investigar, la Oxcarbazepina parece ser más útil para la estabilización del comportamiento que el ácido valproico (Depakine).
No hay comentarios:
Publicar un comentario