Genes del cromosoma 1
Las acil-CoA deshidrogenasas son enzimas que intervienen en la primera de las cuatro reacciones que constituyen la β-oxidación, la principal ruta metabólica de oxidación de los ácidos grasos. En la clasificación de las enzimas corresponde al grupo de lasoxidorreductasas.
Las acil-CoA deshidrogenasas catalizan la deshidrogenación de los carbonos 2 y 3 (α y β, respectivamente) de un acil-CoA graso, con lo que se forma un doble enlace entre dichos carbonos, según la reacción:
 .- .....................................:https://es.wikipedia.org/w/index.php?title=Especial:Libro&bookcmd=download&collection_id=30c0a0131cf020ab05d839b1440105fc4295fd72&writer=rdf2latex&return_to=Acil-CoA+deshidrogenasa
.- .....................................:https://es.wikipedia.org/w/index.php?title=Especial:Libro&bookcmd=download&collection_id=30c0a0131cf020ab05d839b1440105fc4295fd72&writer=rdf2latex&return_to=Acil-CoA+deshidrogenasaDeficiencia de acil-CoA deshidrogenasa de cadena media (MCAD)

La deficiencia de MCAD es un error congénito del metabolismo de los ácidos grasos de cadena media (de 6 a 10 átomos de carbono).
Es el trastorno más frecuente de la oxidación de ácidos grasos, aproximadamente 1:10.000 recién nacidos en raza caucásica, siendo más frecuente en el norte de Europa que en los países mediterráneos.
Causa un bloqueo en la oxidación de ácidos grasos de cadena media (de 6-10 átomos de carbono). Esto hace que, en condiciones de descompensación metabólica, se acumulen dichos ácidos, así como sus derivados conjugados con lacarnitina (acilcarnitinas) y con la glicina (acilglicinas) y ácidos dicarboxílicos en sangre y orina.
¿Cuáles son las manifestaciones clínicas de la deficiencia de MCAD?
Las manifestaciones clínicas de la deficiencia de MCAD se presentan, en general, debido a una descompensación desencadenada por un aumento de las necesidades energéticas del niño.
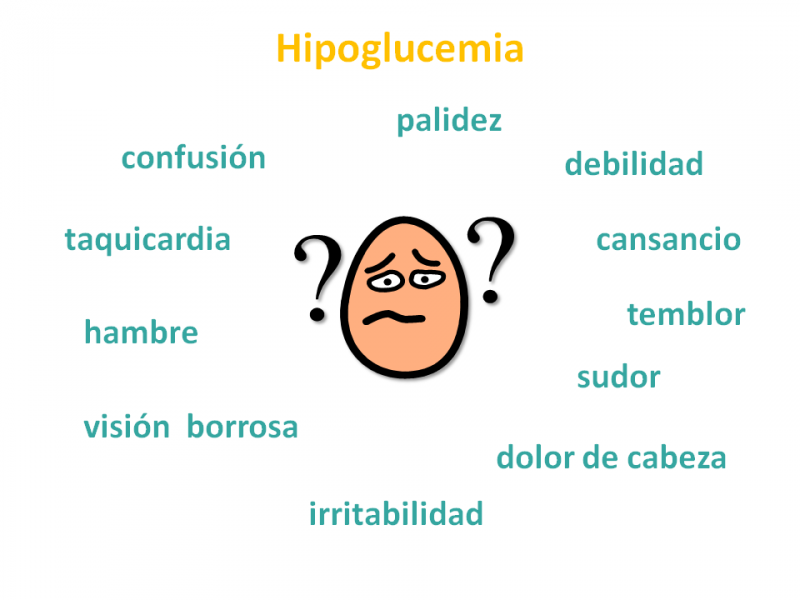
Cuando éstas son superiores al aporte externo de glucosa (a partir de la alimentación) y al aporte interno de glucosa (a partir de la degradación del glucógeno hepático), se pone en marcha la β-oxidación de los ácidos grasos.
Si esta vía está interferida por un defecto de MCAD, se produce una hipoglucemia hipocetósica (con ausencia o deficiencia de cuerpos cetónicos por fallo de síntesis de acetil-CoA, producto final de la beta-oxidación), que puede conducir al coma.
La presentación clínica de la deficiencia de MCAD es muy heterogénea, incluyendo desde pacientes asintomáticos durante muchos años, diagnosticados en estudios familiares o en elcribado neonatal, hasta manifestaciones neonatales de letargia, hipotonía y vómitos, aunque éstas son muy poco frecuentes (> 7% de casos).
La presentación más habitual ocurre antes de los 2 años de vida, entre 10-14 meses, tras un cuadro infeccioso que comporta un aumento de las necesidades energéticas.
Puede presentarse en forma de coma hipoglucémico, episodios de vómitos y letargia, disfunción hepática aguda, síndrome Reye-like, apneas e incluso en algunos casos muerte súbita.
Algunos pacientes pueden presentar episodios recurrentes de descompensación, mientras que otros, una vez diagnosticados y bien controlados, se mantienen asintomáticos.
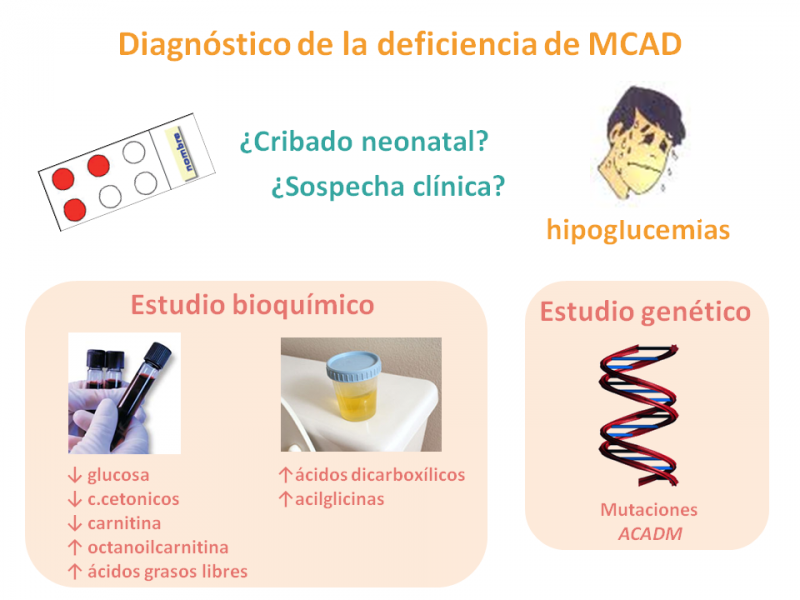
¿Cómo se diagnostica la deficiencia de MCAD?
El diagnóstico se realiza en base al cuadro clínico-bioquímico de hipoglucemia hipocetósica, acompañada a veces de hiperamonemia, o bien mediante el cribado neonatal ampliado.
El análisis de ácidos orgánicos en orina muestra un perfil característico de aciduria dicarboxílica, con hidroxiácidos y derivados de glicina. Los ácidos grasos libres (C8 y C10) en plasma están elevados, así como las acilcarnitinas (octanoilcarnitina) y acilglicinas, mientras que la carnitina libre está deficiente.
El cribado neonatal para la deficiencia de MCAD, con inicio de un tratamiento adecuado, previene muchas de las descompensaciones y sus posibles secuelas, por lo que se está aplicando ya actualmente en muchos países. El cribado neonatal está permitiendo diagnosticar formas leves que tal vez no se presentarían hasta la adolescencia o edad adulta.
El 90% de pacientes caucásicos lleva la mutación A985G en el gen ACADM (el 80% en homocigosis, es decir, los dos alelos de dicho gen con la misma mutación), que afecta a la estabilidad de la proteína enzimática.
Por ello y dada la complejidad del estudio enzimático, se acostumbra a recurrir directamente al estudio genético para confirmar la enfermedad.
El estudio genético permite además el consejo genético familiar y el diagnóstico prenatal, si se requiere.
¿Tiene tratamiento la deficiencia de MCAD?

El tratamiento se basa en prevenir la hipoglucemia, lo que se consigue:
- Evitando el ayuno prolongado, mediante una dieta fraccionada.
- Utilizando una dieta rica en hidratos de carbono, usando hidratos de carbono de absorción lenta.
- Mediante la restricción de grasas de cadena media, por lo que están contraindicados los triglicéridos de cadena media(MCT).
- Ante situaciones de estrés (infecciones, cuadros febriles) evitar ayuno prolongado asegurando una ingesta adecuada de hidratos de carbono (a base de bebidas o alimentos ricos en hidratos de carbono).
Aconsejamos consultar la Pauta de descompensación en un defecto de la β-oxidación y metabolismo de la carnitina.


ACTA1 es un gen de humanos que codifica para la alfa actina 1. La proteína actina codificada por el gen es una de las isoformas típicas el músculo esquelético, formando parte del aparato contráctil del músculo. También se expresa en el músculo cardiaco y en la glándula tiroides.1 Susecuencia consta de siete exones, que producen cinco transcritos conocidos.2
Su expresión génica se induce por los estímulos conducentes a la formación del músculo.3 De hecho, la expresión del gen codificante para la actina provoca la expresión de otros genes miogénicos, esenciales para la formación del músculo.4 Un factor de transcripción implicado en la activación de la expresión de ACTA1 es SRF (el factor de respuesta al suero), una proteína que reconoce y se une su secuencia promotora.5 La actuación de SRF no es única: en muchos casos, otras proteínas intervienen en la activación de la expresión de la actina, como las señales derivadas de la presencia de andrógenos (a veces denominados esteroides anabólicos), lo cual está relacionado con la actividad de estos últimos como inductores del desarrollo muscular.
| Actina, ALPHA, músculo esquelético 1; ACTA1 | |||||||||||||||||
| Títulos alternativos; símbolos | |||||||||||||||||
| ASMA | |||||||||||||||||
| HGNC Aprobado Símbolo Gen: ACTA1 | |||||||||||||||||
| Ubicación citogenético: 1q42.13 coordenadas Genómica (GRCh37): 1: 229,566,991-229,569,857 (de NCBI) | |||||||||||||||||
| Relaciones gen-fenotipo | |||||||||||||||||
| |||||||||||||||||
| TEXTO | |||||||||||||||||
| Descripción | |||||||||||||||||
| El gen codifica ACTA1 músculo esquelético alfa-actina, el principal isoforma de actina en el músculo esquelético adulto, que forma el núcleo del filamento delgado del sarcómero donde interactúa con una variedad de proteínas para producir la fuerza de la contracción muscular ( Laing et al. , 2009 ). | |||||||||||||||||
| Clonación y expresión | |||||||||||||||||
| El uso de ADNc de beta-actina de pollo como sonda, Gunning et al. (1983) clonado alfa-actina de una biblioteca de ADNc de músculo humano.También clonaron beta-actina (ACTB; 102,630 ) y gamma-actina (ACTG1; 102,560 ) a partir de una biblioteca de ADNc de fibroblastos. Análisis de secuencias de los extremos 5-prime reveló que la alfa-actina comienza con tanto una metionina y una cisteína que no se encuentra en la proteína madura. Llegaron a la conclusión de que, ya que las proteínas de actina no conocidas empiezan con una cisteína, debe haber eliminación postraduccional de cisteína además de metionina en la síntesis de alfa-actina, pero no en la síntesis de beta o gamma-actina. Hanauer et al. (1983)clonado alfa-actina a partir de una biblioteca de ADNc a partir de ARNm desarrollado cuádriceps musculares utilizando ADNc de alfa-actina de músculo esquelético de ratón como sonda. La secuencia se caracteriza por un alto contenido de GC (61,6%). Hanauer et al. (1983) observó la conservación de la secuencia de aminoácidos entre actins humanos y de rata, y una comparación de las secuencias codificantes reveló 61% cambios silenciosos. Taylor et al. (1988) clonado alfa-actina y determinó que el transcrito primario codifica una proteína de 377-amino ácido, incluyendo los primeros 2 residuos, que están ausentes de la proteína madura. Señalaron que los mismos 2 codones preceden al codón que especifica el aminoácido N-terminal en los genes homólogos de rata, ratón, pollo, Drosophila, y el erizo de mar. | |||||||||||||||||
| La estructura del gen | |||||||||||||||||
| Taylor et al. (1988) determinaron que el gen de la alfa-actina contiene 7 exones. Hay un gran intrón en la región no traducida 5-prime que es característico de actins y muchos genes específicos de músculo. El promotor contiene una caja TATA y 3 cajas CArG conservadas; Taylor et al.(1988) demostró que éstos fueron activados por la diferenciación de las células musculares en una rata línea celular miogénica. La región no traducida 3-prime contiene una región rica en GC, así como una señal de adición de poli putativo (A). | |||||||||||||||||
| Cartografía | |||||||||||||||||
| Mediante el uso de una sonda de ADNc en híbridos de células somáticas, Hanauer et al. (1984) asignado el gen de la cadena alfa actina de músculo esquelético en el cromosoma 1. secuencias de actina se encontraron en alta rigurosidad también en 2p23-qter y 3pter-q21. En condiciones de baja o media rigurosidad, secuencias de actina se demostraron en la X (p11-p12) y cromosomas Y. Los genes de actina asignados a los cromosomas X e Y (Heilig et al., 1984 ; Koenig et al., 1985 ) parecen ser pseudogenes intronless. Usando una copia de ADNc de la región no traducida 3-prime del gen de alfa-actina de músculo esquelético humano , Muestra et al. (1984) asignan el gen 1p12-qter. Este gen y que para alfa-actina cardíaca (ACTC;102.540 ) se coexpresan tanto en el músculo esquelético y el corazón humano. La coexpresión no es una función de la vinculación; los loci en los cromosomas son separados: (1p21-qter y 15q11-qter, respectivamente . Gunning et al, 1984 ). Usando un panel de híbridos de células somáticas,Alonso et al. (1993) confirmó la localización del gen en el cromosoma humano ACTA1 1. Akkari et al. (1994) redujo la asignación del gen ACTA1 a 1q42 por hibridación in situ fluorescente. También por fluorescencia de hibridación in situ, Ueyama et al. (1995) mapeado el gen para 1q42.1. Sobre la base de análisis de ratón / de hámster híbridos de células somáticas segregantes cromosomas de ratón, Czosnek et al. (1982) concluyeron que el gen de la actina de músculo esquelético se encuentra en el cromosoma del ratón 3. Sin embargo, Alonso et al. (1993) encontró por análisis por PCR de un microsatélite en un retrocruzamiento interespecífico que el gen alfa-actina está estrechamente relacionada con tirosina aminotransferasa y fosforibosiltransferasa adenina en el cromosoma del ratón 8. El gen ACTA1 está situado entre Tat y Aprt; los homólogos humanos TAT ( 613018 ) y Aprt ( 102,600 ) están en el cromosoma humano 16. Abonia et al. (1993) del mismo modo mapeado el gen ACTA1 al cromosoma del ratón 8 por la segregación de RFLVs en 2 juegos retrocruzamiento interespecíficas y en 4 sets ratón endogámicas recombinantes. | |||||||||||||||||
| Función Génica | |||||||||||||||||
| Actina constituye 10 a 20% de la proteína celular y tiene un papel vital en la integridad celular, estructura, y la motilidad. Es altamente conservado durante la evolución. Su función depende del equilibrio entre monomérica (globular) G-actina (42 kD) y (filamentoso) F-actina, un polímero lineal de subunidades G-actina. Entre las proteínas de unión a actina citosólicas, 3 parecen ser de primera importancia en la limitación de la polimerización: profilina ( 176590 , 176610 ), timosina beta-4 ( 300 159 (GSN;), y gelsolin 137.350 ). La existencia de proteínas de unión a actina intracelular permite la concentración de G-actina que se mantiene sustancialmente por encima del umbral en el que normalmente ocurrirá una polimerización y la formación de filamentos. Cuando se libera en el espacio extracelular, la actina, que de otro modo se sabe que tiene un efecto patológico, está obligado por la gelsolina y por la proteína Gc (GC; 139200 ). Este es el llamado sistema actina-scavenger extracelular ( Lee y Galbraith, 1992 ). | |||||||||||||||||
| Características bioquímicas | |||||||||||||||||
| Oda et al. (2009) crearon un modelo de F-actina usando rayos x intensidades de difracción obtenidos a partir de soles de fibra bien orientadas de músculo esquelético de conejo F-actina a 3,3 angstroms en la dirección radial y 5,6 angstroms a lo largo del ecuador. Los autores mostraron que la transición conformacional G- a F-actina es una simple rotación relativa de los 2 dominios principales en alrededor de 20 grados. Como resultado de la rotación de dominio, la molécula de actina en el filamento es plana. La forma plana es esencial para la formación de estable, helicoidal F-actina.Oda et al. (2009) llegó a la conclusión de que su modelo de estructura-F actina sirvió de base para la comprensión de la polimerización de actina, así como sus interacciones moleculares con proteínas de unión a actina. | |||||||||||||||||
| Genética Molecular | |||||||||||||||||
| Resultados de contracción muscular de la fuerza generada entre la delgada proteína actina filamento y el grueso de la miosina proteínas de filamentos, que hace que los filamentos musculares gruesos y finos se deslicen una sobre otra. Hay músculo esquelético, músculo cardíaco, músculo liso, y no musculares isoformas tanto de la actina y la miosina. Las enfermedades hereditarias en los seres humanos se han asociado con defectos en la actina cardíaca en la miocardiopatía dilatada ( 102,540.0001 ) y la miocardiopatía hipertrófica ( 102540.0003 ); en la miosina cardiaca en la miocardiopatía hipertrófica ( 160.760,0001 ); y en la miosina no muscular en la sordera ( 276903.0001 ). En los pacientes con miopatía nemalínica (NEM3; 161.800 ), Nowak et al. (1999) identificaron 15 mutaciones de sentido erróneo diferentes en el gen ACTA1 (véase, por ejemplo, 102610.0001). Las mutaciones de sentido erróneo en ACTA1 se distribuyeron a lo largo de todos los 6 exones de codificación y algunos involucrados dominios funcionales conocidas de actina. Aproximadamente la mitad de los pacientes murió dentro de su primer año, pero 2 pacientes de sexo femenino había sobrevivido en sus treinta años y tuvo hijos. Nowak et al. (1999) identificaron mutaciones dominantes en todo menos en 1 de 14 familias, con las mutaciones sin sentido ser soltero y heterocigotos. La única familia que documenta herencia dominante comprendía una madre afectada de 33 años de edad, con 2 afectadas y 2 niños no afectados ( 102610.0002 ). En otra familia, el padre clínicamente afectado era un mosaico somática para la mutación se observa en sus dos niños afectados. Ellos identificaron mutaciones recesivas en 1 familia en la que los 2 hermanos afectados tenían mutaciones heterocigotas en 2 exones diferentes, 1 paternalmente y el otro de herencia materna ( 102610.0001 ; 102610.0005 ). También identificaron mutaciones de novo en 7 probandos esporádicos para los que fue posible analizar el ADN de los padres. En los miembros afectados de 2 familias con autosómica dominante miopatía "núcleo único", Kaindl et al. (2004) identificaron mutaciones sin sentido en el gen ACTA1 ( 102610.0009 -102610.0010 ). Los pacientes de ambas familias mostraron un curso leve y no progresivo de la debilidad del músculo esquelético. La miopatía fue acompañado por la miocardiopatía hipertrófica del adulto e insuficiencia respiratoria en 1 familia. Histológicamente, se detectaron núcleos en las fibras musculares de al menos 1 paciente de cada familia, mientras que los organismos o varillas nemaline y la acumulación de filamentos de actina estaban ausentes. Kaindl et al. (2004) concluyeron que sus hallazgos establecen mutación en el gen ACTA1 como causa de miopatía congénita dominante con núcleos y delineados otro fenotipo clínico patológica de ACTA1. Por el análisis de inmunoblot, Ilkovski et al. (2004) mostraron que el músculo de miopatía nemalínica (NM) de los pacientes había aumentado los niveles de gamma-filamin (FLNC; 102.565 ), miotilina (TTID; 604.103), desmina (DES; 125.660 ), y alfa-actinina (ACTN1; 102575 ), consistente con la acumulación de cuerpos nemaline derivadas línea Z. No se observaron agregados intranucleares en mioblastos transfección con V163L ( 102610.0004 ) -, V163M-, y R183G-nulos que actúan construcciones transgénicas, y el modelado mostraron estos residuos para ser adyacente a la señal de exportación nuclear de la actina. Transfección estudios mostraron más alteraciones significativas en la capacidad de V136L y R183G actina mutantes para polimerizar y contribuir a los filamentos de actuación insolubles. Los estudios in vitro sugieren que el plegamiento anormal, polimerización alterado, y la agregación de las isoformas de actina mutantes pueden ser propiedades comunes de mutantes ACTA1 NM. Una combinación de estos efectos puede contribuir a las características patológicas comunes de NM, a saber, la formación de intranucleares y citoplasmáticas varilla, la acumulación de filamentos delgados, y la desorganización miofibrilar. Laing et al. (2004) identificaron mutaciones en el gen ACTA1 ( 102610.0011 - 102610.0013 ) en 3 pacientes no relacionados con una forma grave de la congénita de tipo fibra desproporción ( 255.310 ). Ninguno de los pacientes tenía varillas nemaline en el músculo de la biopsia. Laing et al. (2009) proporciona una revisión de mutaciones y polimorfismos en el gen ACTA1 y descrito 85 nuevas mutaciones. Las mutaciones se extienden a lo largo de los 6 exones de codificación, y no hay puntos calientes de mutación. Con independencia de la patología, las mutaciones ACTA1 suelen dar lugar a una miopatía clínicamente grave, con muchos pacientes mueren en los primeros años de vida.La mayoría de las mutaciones son dominantes, y la mayoría de estos son de novo. Cerca de 10% de las mutaciones son recesivas y funcionalmente nulo. | |||||||||||||||||
| Genotipo / fenotipo correlaciones | |||||||||||||||||
| Ilkovski et al. (2001) evaluaron una nueva serie de 35 pacientes con miopatía nemalínica. Se identificaron 5 pacientes no relacionados con una mutación sin sentido en el gen ACTA1 (véase, por ejemplo, 102610.0002 ; 102610,0006 - 102610.0008 ), lo que sugiere que las mutaciones en esta cuenta gen de la enfermedad en aproximadamente el 15% de los pacientes. Los 5 mutaciones eran novedosos, mutaciones de novo dominantes. Un individuo estudiado posteriormente tuvo 2 hijos afectados, un resultado consistente con transmisión autosómica dominante. Los 7 pacientes mostraron una marcada variabilidad clínica, que van desde debilidad congénita aparición severa, con la muerte por insuficiencia respiratoria durante el primer año de vida, a una miopatía leve inicio en la infancia con la supervivencia hasta la edad adulta. Hubo variación marcada tanto en la edad de inicio y la gravedad clínica en los 3 miembros afectados de la familia 1. Las características patológicas compartidos por los pacientes incluidos anormal diferenciación del tipo de fibra, la acumulación de glucógeno, la alteración miofibrilar, y "espirales" de filamentos delgados de actina. El porcentaje de fibras con varillas no se correlacionó con la gravedad clínica; sin embargo, el fenotipo letal severa se asoció con tanto grave, generalizada, la desorganización de la estructura sarcomérica y localización anormal de actina sarcomérica. La marcada variabilidad, en el fenotipo clínico, entre los pacientes con mutaciones diferentes en ACTA1 sugerido que tanto el sitio de la mutación y la naturaleza del cambio de aminoácidos tienen efectos diferenciales sobre la formación de filamento fino y las interacciones proteína-proteína. La variabilidad intrafamiliar sugiere que el genotipo alfa-actina no es el único determinante del fenotipo, sin embargo. En un informe de la conferencia de 2002 sobre la miopatía nemalínica, Wallgren-Pettersson y Laing (2003) señaló que se habían identificado 59 mutaciones en el gen ACTA1. El noventa por ciento de las familias tenían un diagnóstico de miopatía nemalínica, el 11% tenía un diagnóstico de miopatía actina, y el 11% un diagnóstico de miopatía varilla intranucleares. Los hallazgos subrayan la variabilidad fenotípica causada por mutaciones en el gen ACTA1. Entre los pacientes con miopatía nemalínica, la forma grave fue también estuvieron representadas las formas más comunes, pero leves y típicos, y algunos pacientes tenían características asociadas inusuales. La mayoría de los casos fueron esporádicos, pero hubo ejemplos tanto autosómico dominante y herencia autosómica recesiva. No se observaron correlaciones obvias genotipo / fenotipo. Agrawal et al. (2004) encontraron 29 mutaciones ACTA1 en 28 de 109 (aproximadamente 25%) pacientes con miopatía nemalínica. De todo el grupo, las mutaciones ACTA1 fueron responsables de 14 de 25 (56%) de los casos congénitos graves. Diez pacientes con mutaciones ACTA1 tenido la enfermedad típica ", definido como inicio en la infancia o la niñez con hitos retrasados y la supervivencia hasta la edad adulta, y 1 paciente tuvo inicio en la adultez. Cuatro de las familias con mutaciones ACTA1 mostraron herencia autosómica dominante; 1 familia mostró herencia autosómica recesiva; 2 familias sugirieron penetrancia incompleta; los 21 pacientes restantes tenían enfermedad esporádica con mutaciones heterocigotas. La biopsia muscular a las 5 semanas de edad del paciente ACTA1 hereditaria recesiva con enfermedad severa mostró tinción intensa para la actina cardíaca. Agrawal et al. (2004) hizo hincapié en la heterogeneidad fenotípica entre los pacientes con mutaciones ACTA1. Feng y Marston (2009) proporcionan un análisis de mutaciones ACTA1 y llegaron a la conclusión de que no existen patrones funcionales o bioquímicos obvias visto en mutaciones que dan lugar a la misma patología. Aunque algunas mutaciones se predice o han sido demostrado que interfiere con el procesamiento de N-terminal, el plegamiento postraduccional, la polimerización, o la interacción con otras proteínas, a menudo hay desacuerdo en estudios entre la estructura y función de las proteínas mutantes. No hay correlaciones genotipo / fenotipo claras. | |||||||||||||||||
| Modelo Animal | |||||||||||||||||
| Por recombinación homóloga, Crawford et al. (2002) interrumpieron el gen actina esquelético en ratones. Músculos esqueléticos recién nacido de ratones nulos fueron similares a los de los ratones de tipo salvaje en tamaño, tipo de fibra, y la organización ultraestructural. Tanto los animales hemicigotos y homocigotos nulos mostraron un aumento en la actina cardiaca y vascular ( 102620 ) mRNA en el músculo esquelético, sin mRNA actina de músculo esquelético presente en ratones nulos. Los animales nulos parecían normales al nacer y podían respirar, caminar y mamar. Sin embargo, la indemnización prevista por la expresión de actins vasculares y cardíacas era insuficiente para apoyar el crecimiento adecuado del músculo esquelético y / o función. Dentro de 4 días, todos los ratones nulos mostraron un peso corporal significativamente más bajo que sus compañeros de camada normales, y algunos desarrollan escoliosis. Todos los ratones que carecen de actina esquelética murieron en el período neonatal precoz. Ellos mostraron una pérdida de glucógeno y la reducción de la grasa parda, en consonancia con la desnutrición que conduce a la muerte. | |||||||||||||||||
| Las variantes alélicas ( 17 Ejemplos seleccionados ): | |||||||||||||||||
| Table View | |||||||||||||||||
0.0001 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, LEU94PRO [ dbSNP: rs121909519 ] [ ClinVar ] | |||||||||||||||||
| En 2 hermanos infantiles con autosómica recesiva miopatía severa nemalínica-3 (NEM3; 161.800 ) que conducen a la muerte a los 5 y 19 días de edad, respectivamente, Nowak et al. (1999) heterocigosidad identificado compuesto de 2 mutaciones en el gen ACTA1: una transición de T a C en el exón 3, lo que resulta en un (L94P) sustitución leu94-TO-PRO heredado del padre no afectado, y un A-a-G transición en el exón 5, resultando en una glu259-a-val (E259V; 102610.0005 ) sustitución heredado de la madre no afectada. | |||||||||||||||||
0.0002 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, ASN115SER [ dbSNP: rs121909520 ] [ ClinVar ] | |||||||||||||||||
| En una madre y sus 2 hijos que tenían miopatía nemalínica ( 161.800 ), Nowak et al. (1999) identificaron una transición heterocigotos A-a-G en el exón 3 del gen ACTA1, lo que resulta en una sustitución (N115S)-asn115 a ser. Uno de los niños con una forma grave de la enfermedad estaba vivo a los 3 años; la madre y el otro niño tenían formas más leves, y estaban vivos a los 33 y 18 años de edad, respectivamente. Ilkovski et al. (2001)informaron de una mujer de 35 años de edad, con la mutación N115S. Tenía típica miopatía congénita nemalínica con inicio neonatal de dificultades de alimentación, infecciones del tracto respiratorio, hipotonía, diplejía facial y debilidad muscular proximal en las primeras semanas de vida. Su enfermedad era muy lentamente progresiva o no progresiva. Ella tenía un sib más joven afectada y una hija de afectados, de conformidad con herencia autosómica dominante. | |||||||||||||||||
0.0003 MIOPATÍA, actina, congénita, CON EXCESO DE miofilamentos FINAS
| |||||||||||||||||
| ACTA1, GLY15ARG [ dbSNP: rs121909521 ] [ ClinVar ] | |||||||||||||||||
| En el paciente 2 con una miopatía congénita actina (véase 161800 ), reportado por Goebel et al. (1997) , Nowak et al. (1999) identificaron una transversión heterocigótica-G-a C en el exón 2 del gen ACTA1, lo que resulta en un (G15R) sustitución gly15-a-arg. El paciente fue entregado por cesárea de emergencia en 37 semanas de gestación, debido a polihidramnios materno, tenía hipotonía severa que requiere soporte ventilatorio, y murió a la edad de 3 meses. El examen post mortem excluida la atrofia muscular espinal ( 253.300 ). La biopsia muscular mostró grandes áreas de sarcoplasma desprovista de miofibrillas y mitocondrias normales, y se reemplaza con masas densas de filamentos delgados que fueron inmunorreactivas a la actina. Núcleos centrales, barras obvias, desigual fibras rojas y necrosis estaban ausentes. | |||||||||||||||||
0.0004 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, VAL163LEU [ dbSNP: rs121909522 ] [ ClinVar ] | |||||||||||||||||
| En 2 pacientes no relacionados con nemalínica miopatía-3 ( 161.800 ) originalmente reportado por Goebel et al. (1997) , Nowak et al. (1999)identificaron un (V163L) sustitución heterocigótica-val163-A Leu en el gen ACTA1. Sin embargo, la sustitución de aminoácidos fue causada por diferentes cambios de nucleótidos: en un niño todavía vivo en 7,5 años de edad, codón 163 se cambió de GTG (Val) a CTG (Leu); en un niño que murió a los 4 meses de edad, el codón 163 se cambió de GTG (val) para TTG (leu). Un paciente fue hipotónica desde su nacimiento, tenía atrofia de los músculos de la cintura pélvica y escapular, y la miocardiopatía. Él también tenía un paladar ojival. La biopsia muscular mostró regiones subsarcolemales que estaban desprovistos de actividad oxidativa y llenos de densos filamentos delgados de actina-immunopositive. Varillas nemaline intranucleares también estuvieron presentes. El segundo paciente fue hipotónica desde su nacimiento, tenía una cardiomegalia, y murió de insuficiencia cardiorrespiratoria a la edad de 4 meses. La biopsia muscular mostró un predominio de tipo 1 de la fibra, las masas subsarcolemales de filamentos delgados, y las varillas nemaline intranucleares ( Goebel et al., 1997 ). | |||||||||||||||||
0.0005 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, GLU259VAL [ dbSNP: rs121909523 ] [ ClinVar ] | |||||||||||||||||
| Ver 102610.0001 y Nowak et al. (1999) . | |||||||||||||||||
0.0006 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, ILE357LEU [ dbSNP: rs121909524 ] [ ClinVar ] | |||||||||||||||||
| En un niño con miopatía congénita grave nemalínica ( 161.800 ), que murió a la edad de 6 meses de la insuficiencia respiratoria, Ilkovski et al. (2001)identificaron un heterocigoto de novo mutación en el gen ACTA1, lo que resulta en una sustitución (I357L) ile357-a-leu. | |||||||||||||||||
0.0007 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, GLY268CYS [ dbSNP: rs121909525 ] [ ClinVar ] | |||||||||||||||||
| En un paciente con inicio en la infancia de miopatía nemalínica ( 161.800 ), Ilkovski et al. (2001) identificó un (G268C) sustitución heterocigótica gly268-a-cys en el gen ACTA1. | |||||||||||||||||
0.0008 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, ILE136MET [ dbSNP: rs121909526 ] [ ClinVar ] | |||||||||||||||||
| Ilkovski et al. (2001) identificaron un (I136M) sustitución heterocigótica ile136-a-se reunió en el gen ACTA1 en un hombre de 45 años de edad, con miopatía nemalínica ( 161.800 ). Aunque tenía infantil de aparición y el desarrollo motor retardado, su debilidad era no progresiva, y estaba físicamente activo como un adulto y recurre regularmente a larga distancia ciclismo de competición. Tenía una tos débil e infecciones respiratorias frecuentes. La ecocardiografía fue normal. | |||||||||||||||||
0.0009 MIOPATÍA, actina, congénita, con núcleos
| |||||||||||||||||
| ACTA1, ASP3TYR [ dbSNP: rs121909527 ] [ ClinVar ] | |||||||||||||||||
| En 11 miembros afectados de 4 generaciones y 8 hermandades separadas de una familia alemana con miopatía congénita autosómica dominante con núcleos, parte del espectro fenotípico de nemalínica miopatía 3 ( 161.800 ), Kaindl et al. (2004) identificaron un heterocigoto 110G-T transversión en el gen ACTA1, resultando en un (d3y) sustitución ASP3-a-tyr en la proteína madura. | |||||||||||||||||
0.0010 MIOPATÍA, actina, congénita, con núcleos
| |||||||||||||||||
| ACTA1, GLU334ALA [ dbSNP: rs121909528 ] [ ClinVar ] | |||||||||||||||||
| En 5 miembros afectados que abarcan 3 generaciones de una familia china con miopatía congénita autosómica dominante con núcleos, parte del espectro fenotípico de nemalínica miopatía 3 ( 161.800 ), Kaindl et al. (2004) identificaron una transversión 1110A-C en el gen ACTA1, lo que resulta en un (E334A) sustitución glu334-a-ala. Dos miembros de la familia desarrollan cardiomiopatía hipertrófica del adulto e insuficiencia respiratoria. | |||||||||||||||||
0.0011 MIOPATÍA, congénita, CON FIBRA DE TIPO desproporción
| |||||||||||||||||
| ACTA1, ASP292VAL [ dbSNP: rs121909529 ] [ ClinVar ] | |||||||||||||||||
| En un paciente con miopatía de tipo fibra desproporción congénita severa ( 255.310 ), Laing et al. (2004) identificaron una transversión heterocigótica A-a-T en el exón 6 del gen ACTA1, lo que resulta en una sustitución de asp292-a-val (D292V) en una región que forma parte de la superficie de actina monomérica que se expone en el F polímero-actina. La mutación no se identificó en más de 300 cromosomas de control. ADN no estaba disponible desde cualquiera de los familiares del paciente. El uso de la espectrometría de masas y la electroforesis en gel para examinar el músculo esquelético de pacientes, Clarke et al. (2007) determinaron que D292V-actina representó el 50% del total de actina sarcomérica. Los ensayos in vitro mostraron que D292V-actina resultó en disminución de la motilidad debido a las interacciones anormales entre actina y tropomiosina, con tropomiosina estabiliza en la posición "off". Estudios de transfección celulares demostraron que la proteína mutante incorporado en los filamentos de actina y no dio lugar a aumento de la agregación de la actina o interrupción del sarcómero. Clarke et al. (2007) llegó a la conclusión de que las mutaciones ACTA1 resultantes en CFTD causan debilidad al interferir con la función sarcomérica en lugar de estructura. | |||||||||||||||||
0.0012 MIOPATÍA, congénita, CON FIBRA DE TIPO desproporción
| |||||||||||||||||
| ACTA1, LEU221PRO [ dbSNP: rs121909530 ] [ ClinVar ] | |||||||||||||||||
| En un paciente con miopatía de tipo fibra desproporción congénita severa ( 255.310 ), Laing et al. (2004) identificaron una transición heterocigotos-T-a C en el exón 5 del gen ACTA1, lo que resulta en un cambio leu221-TO-PRO (L221P) en una región que forma parte de la superficie de actina monomérica que se expone en el F polímero-actina. La mutación no se identificó en más de 300 cromosomas de control. ADN no estaba disponible desde cualquiera de los familiares del paciente. | |||||||||||||||||
0.0013 MIOPATÍA, congénita, CON FIBRA DE TIPO desproporción
| |||||||||||||||||
| ACTA1, PRO332SER [ dbSNP: rs121909531 ] [ ClinVar ] | |||||||||||||||||
| En un paciente con miopatía de tipo fibra desproporción congénita severa ( 255.310 ), Laing et al. (2004) identificaron una transición heterocigotos C-a-T en el exón 7 del gen ACTA1, lo que resulta en un cambio pro332-a-Ser (P332S) en una región que forma parte de la superficie de actina monomérica que se expone en el F polímero-actina. La mutación no se identificó en más de 300 cromosomas de control. ADN no estaba disponible desde cualquiera de los familiares del paciente. | |||||||||||||||||
0.0014 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, VAL163MET [ dbSNP: rs121909522 ] [ dbSNP: rs121909532 ] [ ClinVar ] | |||||||||||||||||
| En los miembros afectados de una familia con nemalínica miopatía-3 ( 161,800 ) asociado con varillas intranucleares en la biopsia muscular,Hutchinson et al. (2006) identificaron una heterocigotos G-a-Una transición en el exón 4 del gen ACTA1, lo que resulta en un (V163M) sustitución val163-a-met. Otra mutación ha sido reportada en este codón (V163I; 102610.0004 ). Las características clínicas incluyen hipotonía temprano en la vida, debilidad muscular de las extremidades y la atrofia, la cara alta y delgada, y muy arqueado paladar. Biopsias musculares esqueléticas variaron pero tendían a mostrar intranuclear barras dentro de miofibras. Por microscopía electrónica de muestras de músculo de pacientes reportados por Hutchinson et al. (2006) , Domazetovska et al. (2007) encontraron estructura mayormente normales sarcómero con pequeñas áreas de desorganización sarcomérica. Los estudios inmunohistoquímicos mostraron que la mutación V163M resultó en el secuestro de las proteínas de la línea Z del sarcómero y en agregados intranucleares. Hubo algunas pruebas de la regeneración muscular, lo que sugiere un efecto compensatorio.Estudios de cultivos celulares mostraron resultados similares. Transgénicos V161M-mutante Drosophila fueron volador con la desorganización sarcomérica y estructura de la línea Z alterada en el músculo. Los resultados proporcionan un mecanismo para la debilidad muscular. | |||||||||||||||||
0.0015 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, GLU74ASP Y HIS75TYR [ dbSNP: rs267606627 ] [ ClinVar ] | |||||||||||||||||
| En un bebé varón con miopatía grave nemalínica fatal (NEM3; 161.800 ), García-Angarita et al. (2009) heterocigosidad identificado para un alelo portador de 2 mutaciones de novo en cis que afectan a los nucleótidos adyacentes en el exón 3 del gen ACTA1: una transversión 222G-T, lo que resulta en un cambio (E74D) glu74-a-asp, y un 223C-T transición, lo que resulta en un (H75Y) sustitución his75-a-tyr. Ninguno de los padres no afectados realiza cualquiera de las mutaciones, lo que sugiere posible mosaicismo germinal. García-Angarita et al. (2009) señaló que cada mutación previamente se había informado de manera aislada como causante de miopatía nemalínica, pero nunca había sido reportado juntos en el mismo alelo. El fenotipo en su paciente era grave, incluyendo la disminución de los movimientos en el útero, presentación de nalgas, y las contracturas congénitas. Después del nacimiento, no había hipotonía severa, falta de movimientos espontáneos, y la muerte por insuficiencia respiratoria en la edad de 2 meses. Biopsia de músculo esquelético mostró desorganización y nemaline varillas miofibrilares. | |||||||||||||||||
0.0016 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, LYS328ASN [ dbSNP: rs398122936 ] [ ClinVar ] | |||||||||||||||||
| En un lactante con nemalínica miopatía-3 ( 161,800 ) que se presentó con un fenotipo atípico de la rigidez y la hipertonicidad, Jain et al. (2012)identificaron una transversión 984G-C heterocigótica de novo en el gen ACTA1, resultando en una lys328-a-Asn (K328N) sustitución (K326N en la proteína madura). La biopsia del paciente mostró cuerpos nemaline y 32% de actina mutante. En el análisis de la motilidad in vitro de filamentos delgados de actina derivadas de tejido de la paciente mostró aumento de la sensibilidad al calcio, lo que indica un estado activado. Expresión de los mutantes en las células musculares de ratón no dio lugar a la formación de estructuras similares a varillas, lo que sugiere un mecanismo diferente de la formación de cuerpo nemalínica. El tratamiento médico es ineficaz, y el paciente murió a la edad de 9 meses en un episodio en asistolia. El informe ampliado el espectro fenotípico asociado con mutaciones ACTA1 para incluir la rigidez, la rigidez y la hipertonicidad. | |||||||||||||||||
0.0017 nemalínica MIOPATÍA 3
| |||||||||||||||||
| ACTA1, TRP358CYS [ dbSNP: rs587777354 ] [ ClinVar ] | |||||||||||||||||
| En un niño japonés de 9 años de edad con NEM3 ( 161.800 ) que desarrolló una miocardiopatía dilatada fatal, Gatayama et al. (2013) identificaron un heterocigoto c.1074G-T transversión en el exón 7 del gen ACTA1, lo que resulta en un (W358C) sustitución trp358-to-cys. La mutación no se encontró en 50 controles japoneses. No se realizaron estudios funcionales de la variante. El paciente tenía el desarrollo motor normal, en la primera infancia, pero mostró debilidad muscular leve no progresiva del esqueleto, como el frenado corriendo en comparación con sus pares. Otras características incluyen hipotonía, facies miopáticos, paladar ojival, y debilidad leve de músculos proximales y distales. Presentó a la edad de 9 años con deterioro agudo de la función cardiaca, y murió de insuficiencia cardiaca 6 meses después. El examen post mortem del músculo cardíaco mostró variación en el tamaño de la fibra miocárdica y algunas estructuras finas electrodensos relacionados con líneas Z. Biopsia muscular esquelético había demostrado previamente barras típicas nemaline. Gatayama et al. (2013) señaló que la miocardiopatía dilatada de inicio infantil es poco frecuente en los pacientes con mutaciones ACTA1. |
No hay comentarios:
Publicar un comentario