| Lesión de Bankart | |
|---|---|
 | |
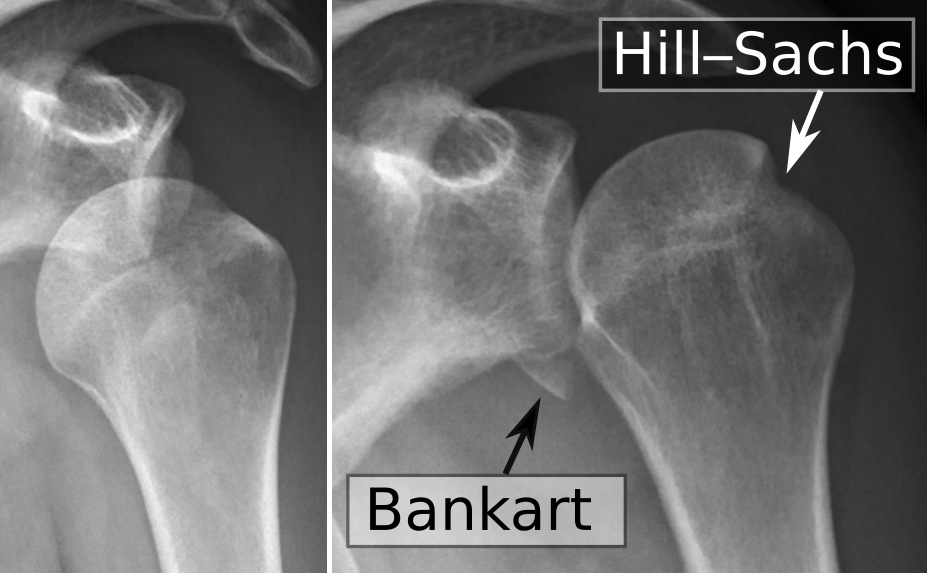
| El labrum glenoideo , el ligamento glenoideoetiquetado , está dañado en una lesión de Bankart .Se muestra una vista lateral que muestra la superficie articular de la escápula derecha. |
Una lesión de Bankart es una lesión del labrum glenoideo anterior ( inferior ) del hombro debido a la dislocaciónanterior del hombro . [1] Cuando esto sucede, se forma un bolsillo en la parte frontal de la glenoides que permite que la cabeza humeral se disluya en ella. Es una indicación para la cirugía y, a menudo, se acompaña de una lesión de Hill-Sachs , daño en la cabeza humeral posterior . [2]
La lesión de Bankart lleva el nombre del cirujano ortopédico inglés Arthur Sydney Blundell Bankart (1879–1951). [3]
Un Bankart óseo es una lesión de Bankart que incluye una fractura en la cavidad glenoidea anterior-inferior del huesode la escápula .
Diagnóstico [ editar ]
El diagnóstico generalmente se realiza inicialmente mediante una combinación de examen físico e imagenología médica , donde esta última puede ser una radiografía de proyección (en casos de Bankart ósea) y / o MRI del hombro. La presencia de contraste intraarticular permite una mejor evaluación del labrum glenoideo . [5] Las roturas tipo V SLAP se extienden al defecto de Bankart. [6]
Tratamiento [ editar ]
La reparación artroscópica de las lesiones de Bankart tiene buenas tasas de éxito, aunque casi un tercio de los pacientes requieren cirugía adicional para continuar con la inestabilidad después del procedimiento inicial en un estudio de adultos jóvenes, con tasas de reoperación más altas en los menores de 20 años. [7] Las opciones de reparación incluyen una técnica artroscópica o un procedimiento Latarjet abierto más invasivo , [8] con la técnica abierta que tiende a tener una menor incidencia de dislocación recurrente, pero también un rango de movimiento reducido después de la cirugía.


| Síndrome de Bannayan – Riley – Ruvalcaba | |
|---|---|
 | |
| Autosómico dominante es la manera en que se hereda esta condición. | |
| Especialidad | Oncología , genética médica |
| Causas | Mutaciones en el gen PTEN[1] |
| Metodo de diagnostico | Basado en signos y síntomas[2] |
| Tratamiento | Basado en los síntomas [2] |
El síndrome de Bannayan-Riley-Ruvalcaba ( BRRS, por sus siglas en inglés ) es un raro síndrome de sobrecrecimiento y un trastorno hamartomatoso con aparición de lipomas subcutáneos múltiples , macrocefalia y hemangiomas . La enfermedad se hereda de manera autosómica dominante . [3] La enfermedad pertenece a una familia de síndromes de poliposis hamartomatosa, que también incluye el síndrome de Peutz-Jeghers , poliposis juvenil y síndrome de Cowden . La mutación del gen PTENsubyace a este síndrome, así como al síndrome de Cowden,Síndrome de Proteus , y síndrome de Proteus-como , estos cuatro síndromes se refieren como PTEN Síndromes Hamartoma-tumorales.
Signos y síntomas [ editar ]
El síndrome de Bannayan-Riley-Ruvalcaba se asocia con cabeza agrandada y hamartomas mesodérmicos benignos ( hemangiomas múltiples y pólipos intestinales ). También puede haber disemorfia y retraso en el desarrollo neuropsicomotor. [5] [4] El agrandamiento de la cabeza no causa el ensanchamiento de los ventrículos ni la presión intracraneal elevada ; estos individuos tienen un mayor riesgo de desarrollar tumores , ya que el gen involucrado en los BRR es el homólogo a la fosfatasa y la tensina. [ cita médica necesaria ]
Algunas personas tienen problemas de tiroides en consonancia con bocio multinodular , adenoma de tiroides , no diferenciada medular cáncer de tiroides , la mayoría de las lesiones están creciendo lentamente. La afectación visceral e intracraneal puede ocurrir en algunos casos y puede causar sangrado y compresión mecánica sintomática [6] [7]
Genética [ editar ]

La genética del síndrome de Bannayan-Riley-Ruvalcaba se determina, en la mayoría de los casos, a través del gen PTEN que presenta alrededor de 30 mutaciones en esta afección. Este gen que regula el crecimiento celular , cuando no funciona correctamente puede conducir a hamartomas. La ubicación cromosómica de PTEN es 10q23.31, mientras que la ubicación molecular es de 87.863.438 a 87.971.930 [1] [7]Hay muchos síndromes que están vinculados a PTEN además del síndrome de Bannayan-Riley-Ruvalcaba. [8]
El síndrome combina el síndrome de Bannayan-Zonana, el síndrome de Riley-Smith y el síndrome de Ruvalcaba-Myhre-Smith. [9] El síndrome de Bannayan-Zonana se llama así por George A. Bannayan y Jonathan Zonana [10]
Diagnóstico [ editar ]
En términos de diagnóstico del síndrome de Bannayan-Riley-Ruvalcaba, no existe un método actual fuera de las características físicas que puedan presentarse como signos / síntomas. [2] Sin embargo, existen pruebas genéticas moleculares múltiples (y pruebas citogenéticas ) para determinar el síndrome de Bannayan-Riley-Ruvalcaba. [11]
El diagnóstico diferencial [ editar ]
El diagnóstico diferencial para BRRS consiste en lo siguiente: [12]
Tratamiento [ editar ]

En términos de tratamiento / manejo, se debe observar qué signos o síntomas están presentes y, por lo tanto, tratarlos como no hay ninguna otra guía actual. El individuo afectado debe ser monitoreado para detectar cáncer de: [2]
No hay comentarios:
Publicar un comentario