Bioquímica clínica
La farmacocinética es la rama de la farmacología que estudia los procesos a los que unfármaco es sometido a través de su paso por el organismo. Trata de dilucidar qué sucede con un fármaco desde el momento en el que es administrado hasta su total eliminación del cuerpo.
Para ello, se han desarrollado diferentes modelos que simplifiquen los numerosos procesos que tienen lugar entre el organismo y el fármaco. Aún cuando dentro de los mismos elmodelo policompartimental es el más próximo a la realidad, la complicación que conlleva ha hecho que sean los modelos monocompartimental y en todo caso elbicompartimental los más usados. Desde esos prismas, el estudio detallado de los sucesivos pasos que atraviesa el fármaco en el organismo, se agrupan con el acrónimoLADME:
- Liberación del producto activo,
- Absorción del mismo,
- Distribución por el organismo,
- Metabolismo o inactivación, al ser reconocido por el organismo como una sustancia extraña al mismo, y
- Excreción del fármaco o los residuos que queden del mismo.
Estas distintas fases, implican la utilización y manejo de conceptos básicos para comprender la dinámica instaurada. Así, las propiedades de las sustancias que actúan como excipientes, las características de las membranas biológicas y la forma en que las sustancias pueden atravesarlas, o las características de las reacciones enzimáticas que inactivan al fármaco, son de necesario conocimiento para la correcta comprensión de la cinética del fármaco.- ..................................................:http://es.wikipedia.org/w/index.php?title=Especial:Libro&bookcmd=download&collection_id=94e1cb260eaccf65783e3620f9f5ca72ede60d03&writer=rdf2latex&return_to=Farmacocin%C3%A9tica
FARMACOCINÉTICA
La farmacocinética es la ciencia que estudia el paso de los fármacos a través del cuerpo. Es la velocidad del paso del fármaco por dentro del cuerpo. Ve el efecto que tiene el organismo bajo los fármacos.
Los organismos tienden a eliminar xenobióticos fuera del organismo. La farmacocinética engloba o refleja todos los procesos LADME.
El comportamiento farmacocinético será el reflejo de estos procesos. Las dosis de fármaco cuando se administran se hacen en función del peso del animal.
En humano se calculan las dosis para pesos medios de 60 Kg y después sólo se modifica en los niños pequeños.
En los animales, se tiene que ajustar las dosis en función del peso del animal. Normalmente siempre se dan las dosis en:
DOSIS = mg, mg, g (F)
Kg peso animal
Ej: 50 mg / Kg à 4 Kg peso à 4 x 50 = 200 mg Fármaco
à 20 Kg peso à 50 x 20 = 1000 mg Fármaco.
Algunos de los cálculos que se hacen para estudiar la farmacocinética se hacen por Kg de peso. La herramienta que se usa para estudiar la farmacocinética es la sangre. Se administra el fármaco y se saca sangre para determinar las dosis de fármaco en sangre. Se llaman curvas de niveles plasmáticos. Normalmente los análisis se hacen con plasma (después de centrifugar).
Se toman muestras separadas a lo largo del tiempo:
Incremento del fármaco
| ||||
10 min -> C1
|
30 min -> C3
|
2 h -> C5
| ||
20 min -> C2
|
60 min -> C4
|
4 h -> C6
| ||
Según la vía en la que se administre un fármaco, pueden haber diferentes niveles de concentraciones plasmáticas. Si se administra IV, el fármaco va directamente a la sangre. El fármaco sólo se irá eliminando porque directamente aumenta la concentración.
Si se administra el fármaco por otra vía donde haya una absorción, tendremos la concentración máxima en sangre cuando el fármaco se haya absorbido del todo. Desde el momento en que se administra, el fármaco se empieza a absorber, llega al máximo y después se comienza a eliminar.
En función de las vías de administración y de la velocidad que la que se absorbe el fármaco hay diferentes curvas. La que tiene más velocidad es la que llega antes a la concentración máxima en el mínimo tiempo posible. A la hora de calcular el comportamiento farmacocinético, se usan las curvas de nivel plasmático.
Después se hacen análisis que permiten decir cuanto tiempo se mantiene el fármaco en el organismo, la concentración máxima en la sangre, en qué concentraciones se encuentra el fármaco en la sangre, si llega al lugar de acción, si el fármaco se distribuye por los tejidos periféricos...
Se pueden establecer las dosis adecuadas y cada cuanto tiempo se tiene que establecer esta dosis. Las curvas del nivel plasmático son el resultado de los procesos LADME. Estos procesos pueden ser de diferente tipo según su naturaleza. Cada uno de ellos tiene un proceso cinético de un orden determinado.
PROCESOS CINÉTICOS
q Proceso cinético de orden 1 à si se administra una cantidad de fármaco al organismo por la vía IV, el fármaco se disuelve en el organismo. Después, el fármaco se va eliminando. La velocidad de desaparición es la variación de esta cantidad en función del tiempo y es una función de una constante y de la cantidad de fármaco que hay.
La velocidad con la que la cantidad de fármaco disminuye dentro de la sangre depende de la cantidad de fármaco que hay en el organismo, a medida que va disminuyendo la cantidad de fármaco porque va disminuyendo de la sangre, la cantidad de filtrado será menor. La velocidad a la que se elimina depende de la velocidad de la sangre. Es un proceso de orden 1. la variación de la cantidad de fármaco que hay en el organismo en función del tiempo depende de una constante y de la cantidad de fármaco que hay en el organismo. Estos procesos se encuentran en la absorción, distribución y metabolismo.
q Proceso cinético de orden 0 à la velocidad de desaparición del fármaco de la sangre es constante. Siempre desaparece el fármaco a la misma velocidad. Ej: procesos que funcionan con un transportador.
Si el fármaco no se metaboliza y se elimina por la vía renal con un transportador, indica que si el transportador está saturado, sólo se elimina la cantidad de fármaco correspondiente al número de transportadores.
t0 = 0 à 20 mg
|
t1 = 1 à 18 mg
|
t2 = 3 à 16 mg
|
t3 = 4 à 14 mg
|
La eliminación es independiente de la cantidad de fármaco en la sangre.
q Proceso cinético de orden mixto à aquellos procesos en los que intervienen transportadores.
Ej: sistema de transportadores que después de administrar la dosis, va disminuyendo. La eliminación es lineal. Cuando la cantidad que hay en sangre es inferior al número de transportadores, el sistema deja de estar saturado. Se transforma en un proceso cinético de orden 1 y la eliminación depende de la cantidad de fármaco que hay en sangre. Estos procesos son las cinéticas de Michaellis-Menten, en las cuales, si representamos la velocidad de la reacción o eliminación en función de la cantidad de fármaco en sangre:
La variación de la cantidad en función del tiempo (velocidad) es igual a la velocidad máxima por la cantidad de fármaco que hay en un momento dado, partido por la Km y la cantidad de fármaco que hay en el sistema.
La Km es la cantidad de fármaco cuando la velocidad es la mitad de la velocidad máxima.
Las curvas del nivel plasmático que se obtienen son el conjunto de todos los procesos LADME. Una absorción de orden 0 (mediante transportadores) puede quedar enmascaradas por los procesos de eliminación, metabolismo... de orden 1 y puede alterar el proceso cinético.
Si se administra un fármaco que se metaboliza por la vía IV, se ve una cinética de cantidad dependiente porque afecta a la excreción y el metabolismo de una forma que no vemos.
MODELOS FARMACOCINÉTICOS
Normalmente el primer estudio farmacocinético de un fármaco es IV porque sólo hay distribución, metabolismo y excreción. La absorción del fármaco puede modificar mucho el estudio.
Hay diferentes tipos de modelos farmacocinéticos en función de las curvas que se obtienen en la administración IV.
§ Modelo monocompartimiental à se asume que después de la administración de la dosis de forma IV, el fármaco se distribuye inmediatamente en un compartimiento. Este compartimiento se comporta como si fuera acuoso. Si suponemos que el fármaco después de administrarse sólo va a sangre y no puede superar el endotelio capilar, el fármaco sólo se encuentra en la sangre. Si el fármaco puede atravesar el endotelio capilar y llegar al líquido intersticial, el compartimiento es la sangre y el líquido intersticial. Si el fármaco puede distribuirse uniformemente por todos los tejidos del organismo inmediatamente, el comportamiento de este organismo es todo el organismo. Como el fármaco se distribuye uniformemente por todo este compartimiento, la concentración de fármaco en este compartimiento siempre será la misma.
La concentración del líquido intersticial es la misma que la concentración del plasma porque la distribución es homogénea. Da como resultado que se administra la dosis en el compartimiento. El compartimiento tiene una cantidad de fármaco, dosis y volumen. Esta dosis se distribuye uniformemente por el compartimiento y se comienza a eliminar por orden 1, orden 0 u orden mixta.
Generalmente los fármacos se eliminan por las vías de orden 1 porque
dQ / dt = -kQ
Esta k es la constante de eliminación. Desde que se administra el fármaco en adelante, hay una cantidad de fármaco remanente y una cantidad de fármaco eliminada.
dC / dt = - ke C
Generalmente nosotros sólo podemos fijarnos en la sangre. Se tiene que interpretar las curvas y decidir el comportamiento del fármaco.
§ Modelo bicompartimiental à refleja que el fármaco es distribuido en dos compartimientos. Cuando se administra el fármaco, un aparte se distribuye directamente y de forma rápida (tejido compartimiento central) y otra se distribuye más lentamente (tejidos periféricos).
La velocidad de entrada en el compartimiento periférico es bastante lenta. Pasado cierto tiempo, la cantidad de fármaco del compartimiento periférico es suficientemente grande para que la entrada sea igual a la salida. El intercambio entre el compartimiento central y el periférico es como si no estuviera, porque la entrada y salida son iguales y la curva sólo indicará la eliminación.
La velocidad de eliminación del fármaco está en función de la constante de eliminación y de la cantidad de fármaco del compartimiento central más la constante de entrada al compartimiento periférico por la cantidad de fármaco del compartimiento central más la constante de entrada del fármaco del compartimiento periférico al central por la cantidad de fármaco en el compartimiento periférico.
dQ / dt = -keQ – k12Qc + k21Qp
Así se puede ajustar cada vez más nuestros puntos experimentales a unos modelos para que nos represente nuestras formas.
Bioquímica clínica
En farmacología, la farmacodinámica o farmacodinamia, es el estudio de los efectos bioquímicos y fisiológicos de los fármacos y de sus mecanismos de acción y la relación entre la concentración del fármaco y el efecto de éste sobre un organismo. Dicho de otra manera: el estudio de lo que le sucede al organismo por la acción de un fármaco. Desde este punto de vista es opuesto a lo que implica la farmacocinética: a lo que un fármaco es sometido a través de su paso por el organismo.
La farmacodinámica puede ser estudiada a diferentes niveles, es decir, sub-molecular, molecular, celular, a nivel de tejidos y órganos y a nivel del cuerpo entero, usando técnicas in vivo, post-mortem o in vitro.- ...................................................................:http://es.wikipedia.org/w/index.php?title=Especial:Libro&bookcmd=download&collection_id=334683f0151becf1f349623e70183f7734e86ba4&writer=rdf2latex&return_to=Farmacodin%C3%A1mica
FARMACOCINÉTICA
La farmacocinética define la relación que se establece entre el antimicrobiano y el paciente, cómo el organismo manipula la droga, e incluye los procesos de absorción, distribución, unión a proteínas séricas e hísticas, metabolismo y eliminación.3,4 Diferencias en el grado de unión a proteínas séricas pueden originar cambios en la concentración de antibacteriano libre, determinante de la penetración a tejidos y la actividad antibiótica.3
ABSORCIÓN
La absorción de la droga en la circulación sistémica ocurre desde cualquier sitio en el que sea administrada, excepto cuando se administra directamente en un compartimiento fluido fisiológico (fluido cerebrospinal o torrente sanguíneo) donde evidentemente la infusión es directa; esta definición incluye la vía intramuscular, subcutánea, tópica y gastrointestinal después de la administración por vía oral, rectal u otra vía que la lleve al tracto gastrointestinal. La velocidad y el grado de absorción es altamente dependiente de las propiedades físico-químicas de la droga así como del ambiente en el sitio de administración. Propiedades como tamaño molecular, solubilidad, lipofilicidad, estabilidad, influencian la velocidad y extensión de la absorción.5 La cantidad del antimicrobiano que alcanza la circulación sistémica se expresa en porcentaje de la cantidad total que pudo ser adsorbida; este porcentaje se define como biodisponibilidad de la droga.4 Los medicamentos que se adsorben desde el intestino delgado son afectados por el efecto de primer paso por el hígado a través de la circulación portal; drogas administradas por la vía intravenosa, e intramuscular no son afectadas por este fenómeno y tienen mayor biodisponibilidad. La disminución en la perfusión gastrointestinal, subcutánea y muscular que se observa en pacientes sépticos, puede reducir significativamente la absorción de fármacos4,6,7 y por tanto, su biodisponibilidad, generando concentraciones plasmáticas insuficientes de antimicrobianos; en estos pacientes debe priorizarse la administración por vía intravenosa en cuyo caso se alcanza 100 % de absorción.
DISTRIBUCIÓN
La distribución de una droga, es descrita más comúnmente por su volumen de distribución (Vd), el cual no es un volumen real, es un parámetro cinético, permite relacionar la cantidad en el organismo con la concentración en este, es un espacio de dilución.8 Puede definirse como el volumen que debería tener el organismo para que la cantidad presente al equilibrio estuviera a la misma concentración; existen factores que afectan Vd como solubilidad lipídica, coeficiente de partición de la droga entre diferentes tipos de tejidos, flujo sanguíneo en el tejido, pH, y unión a material biológico (ej., proteínas plasmáticas, componentes celulares);4,5 el Vd se puede calcular de la siguiente manera:
El Vd es variable entre personas, se afecta por factores como, disfunción de órganos excretores u obesidad, y puede tener severas variaciones en un mismo individuo, como consecuencia del aumento de la permeabilidad vascular que acompaña a enfermedades graves, sepsis, quemaduras, cirrosis hepática, insuficiencia cardiaca, renal,4 etc, esto agravado por el aporte masivo de fluidos.5,9,10
Es importante para el clínico el conocimiento del concepto Vd; las drogas con pequeños Vd tienen limitada distribución, mientras que las de grandes Vd se distribuyen extensamente por todo el organismo; de este modo se puede inferir que antimicrobiano, se restringe al espacio intravascular y extravascular, lo cual implica en su distribución el medio intracelular.11,12
El ritmo con que la droga se mueve de la sangre al tejido se describe como aclaramiento distribucional; este describe el volumen de sangre del cual el medicamento es transferido al tejido por unidad de tiempo, el aclaramiento distribuciónal es un proceso bidireccional que refleja el equilibrio en el movimiento de la sangre al tejido y del tejido a la sangre.11
METABOLISMO Y BIOTRANSFORMACIÓN
Los antimicrobianos son metabolizados por reacciones que ocurren en el hígado y en otros órganos. Las reacciones metabólicas son clasificadas como de fase I y fase II. Las reacciones de fase I pueden inactivar, activar o convertir un sustrato activo en otro activo, con mayor, menor o igual actividad; estas reacciones están controladas por el sistema del citocromo P-450; generalmente inactivan al sustrato, lo hacen más polares, lo que facilita su eliminación. Las reacciones de fase II son procesos en los que interviene la conjugación de estos compuestos con grandes moléculas, incluye glucoronidación, sulfatación y acetilación, esto aumenta la polaridad y facilita la excreción.3-5,11
ELIMINACIÓN
La eliminación de sustancias extrañas ocurre por 2 mecanismos fundamentales de excreción, aclaramiento renal(CLr) descrito como el ritmo con que es eliminada una sustancia del organismo a través de los riñones, incluye filtración glomerular, secreción tubular, y difusión pasiva; diferentes antimicrobianos son eliminados por uno o más de estos procesos.4
Aclaramiento no renal, es un término genérico que describe la suma de vías de aclaramiento que no incluyen al riñón; estos mecanismos incluyen vías biliares (ej., ceftriaxone) o intestino (ej., azitromicina); otros mecanismos menos comunes como la eliminación del alcohol a través de la piel y pulmones y la ionización e inactivación de los aminoglucósidos por el esputo y eliminación por la expectoración en fibrosis quística.
El ritmo de aclaramiento renal y no renal se combinan para determinar el ritmo con que es eliminado el antimicrobiano del organismo, esto se conoce como aclaramiento corporal total.11 Del concepto de eliminación deriva el de tiempo de vida media del antimicrobiano (t1/2), la vida media de la droga es el tiempo que se requiere para que la concentración en sangre del compuesto decrezca a la mitad.4 Se considera que la estabilidad en la concentración de una droga se alcanza cuando el paciente la ha estado tomando por un periodo igual a 5 o 7 veces su t1/2 (ej., 5 a 7 días para una droga con una vida media de 24 h); de manera similar se considera que se ha eliminado cuando desde la última dosis ha pasado un tiempo similar a 5 o 7 veces su t1/2; la vida media de una droga varía de paciente a paciente, en ocasiones se reporta como rangos; la unión a proteína y estados de fallos finales de órganos alteran la t1/2 de una droga, Ej. la eliminación de antimicrobianos como beta-lactámicos, vancomicina, aminoglucósidos y quinolonas puede reducirse significativamente en casos de insuficiencia renal, se generan concentraciones plasmáticas más elevadas; en el caso de drogas con margen terapéutico estrecho como aminoglucósidos, puede dar origen a toxicidad, la dosificación de estos fármacos debe ajustarse en forma proporcional a la función renal; los antimicrobianos de eliminación hepática como lincosaminas y antituberculosos deben ajustarse en la disfunción hepática; sin embargo, el aclaramiento de fármacos en casos de insuficiencia hepática es mucho más difícil de estimar. Los antibacterianos que tienen eliminación mixta, como cloxacilina o ceftriaxona, en general no requieren ajuste de dosis frente a la falla de un órgano excretor, por un aumento compensatorio de la depuración por el otro órgano.
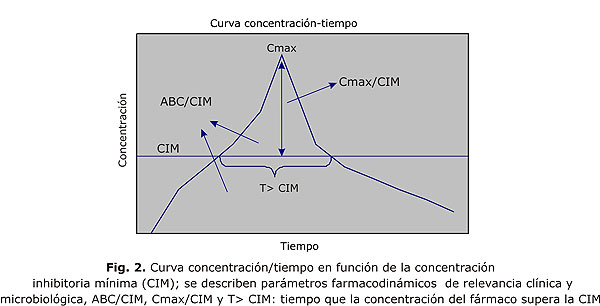
Las características de absorción, distribución y eliminación del antimicrobiano determinan la curva concentración/tiempo en plasma, de esta se infiere la concentración que alcanza el fármaco en el tejido infectado, donde, en definitiva se requiere de este en concentraciones adecuadas para el control de la infección. Esta curva define una serie de parámetros farmacocinéticos importantes, concentración máxima (Cmáx), la vida media del antimicrobiano en el plasma (t1/2) y el área bajo la curva (ABC) (fig. 1).

Las concentraciones plasmáticas e hísticas no siempre se correlacionan linealmente, los antibacterianos con menos unión a proteínas como aminoglucósidos y quinolonas, tienen generalmente una correlación plasma/tejidos mayor que los beta-lactámicos que se caracterizan por una elevada unión a proteínas plasmáticas. Ciertas afecciones como meningitis pueden mejorar transitoriamente la penetración de antimicrobianos al sitio infectado, al aumentar la permeabilidad de la barrera hematoencefálica; en general, la penetración a tejidos es relevante en infecciones que afectan órganos con baja penetración de antibacteriano como son el SNC, ojo, huesos, páncreas y pulmón
En términos generales, para que un antimicrobiano sea efectivo, debe lograr concentraciones superiores a la concentración inhibitoria mínima del microorganismo (CIM), o sea, para que una bacteria se considere susceptible tiene que tener una CIM alcanzable por el antimicrobiano en su perfil farmacocinético en humanos. La farmacodinámica describe el efecto antimicrobiano del medicamento en el sitio de la infección y también los efectos tóxicos en relación con la concentración de la droga durante la evolución de la terapia.13,14 Los estudios farmacodinámicos se basan en 2 tipos de datos, microbiológicos y farmacocinéticos. La farmacodinámica describe la compleja interrelación que se establece entre el perfil farmacocinético del antimicrobiano y la susceptibilidad in vitro de la bacteria; la curva concentración/tiempo del antimicrobiano se determina en función de CIM (fig. 2). Los parámetros farmacocinéticos son expresados en función de la CIM. El éxito clínico y microbiológico depende de una adecuada interacción farmacodinámica entre el antimicrobiano y la bacteria, lo que permite establecer ciertos objetivos farmacodinámicos en el tratamiento antiinfeccioso, como Cmáx/CIM, ABC/CIM o T> CIM que constituyen demostradamente parámetros predictores de éxito.15
CONCENTRACIÓN DE PREVENCIÓN DE MUTANTES
Concentración de prevención de mutantes (CPM) es un concepto que cada vez es más discutido en la literatura, se refiere a la concentración del antimicrobiano que previene la aparición de mutaciones que conducen a resistencia,16 esta varía para diferentes organismos y para diferentes drogas. De este concepto deriva el de ventana de selección de mutantes, se define como el periodo de exposición del germen al antimicrobiano que se caracteriza por una concentración por debajo de CPM pero por encima de la CIM17-19 (fig. 3); teóricamente en este periodo pueden aparecer gérmenes resistentes al antimicrobiano, obtener niveles superiores a CPM de la droga sería un objetivo durante el tratamiento para evitar la selección de cepas mutantes resistentes.

{kind=link}
EFECTO POSANTIBIÓTICO
Durante el testage de antimicrobianos in vitro, se comprobó que al retirar el medicamento existe una demora para que el microorganismo reentre en el periodo logarítmico de crecimiento, este fenómeno se denomina efecto posantibiótico (EPA); la duración de este es dependiente del germen y la droga. Aminoglucosidos, fluoroquinolonas, eritromicina, clindamicina, tetraciclinas y estreptograminias,20 producen in vivo EPA contra organismos gramnegativo, los beta-lactámicos (excepto carbapenémicos) no producen EPA contra organismos gramnegativos;21,22 los beta-lactámicos producen breve EPA contra organismos grampositivos, in vivofundamentalmente contra estafilococos. De manera general, los antimicrobianos que interfieren con la síntesis proteica o del ADN poseen EPA contra gérmenes gramnegativo y los que interfieren en la síntesis de pared no. Del fenómeno EPA depende el efecto post-antibiótico sub-CIM, se refiere a que el microorganismo en fase de retardo de recrecimiento por el efecto post-antibiótico es más susceptible a reducir su crecimiento a concentraciones sub-CIM y el efecto leucocitario posantibiótico, define que el microorganismo en el estado post-antibiótico de crecimiento es más susceptible a la actividad microbicida leucocitaria. Ambos efectos ocurren in vivo donde la concentración de la droga declina gradualmente en el tiempo. Los mecanismos del EPA son desconocidos, posibles explicaciones incluyen daños no letales inducidos por el medicamento y persistencia del antimicrobiano en el sitio de acción;23 nuevos microorganismos inyectados durante el periodo de EPA, inician un rápido crecimiento sugiriendo que el EPA no es causado por la persistencia de la droga en el tejido. En el orden clínico la presencia o ausencia del EPA se puede usar para variar los esquemas de dosis de los antimicrobianos, un agente con un prolongado EPA se puede usar con menos frecuencia que uno que no posea EPA. Alternativamente, un agente que no posea EPA puede ser más efectivo en infusión continua, de manera que las concentraciones séricas siempre excedan la CIM del germen en cuestión.
El mecanismo de acción de cada familia de antimicrobianos determina una cinética bactericida específica, y la presencia y duración del efecto post-antibiótico. De esta manera se pueden dividir los antimicrobianos en 3 grupos:
• El primer grupo induce muerte concentración dependiente y produce EPA moderado o prolongado. • En el segundo grupo el patrón de destrucción es dependiente del tiempo de exposición y no produce o tienen un mínimo EPA contra la mayoría de los microorganismos. • El tercer grupo destruye de manera tiempo dependiente pero difiere del grupo anterior en que poseen EPA moderado o prolongado.
AGENTES QUE DESTRUYEN BACTERIAS DE MANERA CONCENTRACIÓN DEPENDIENTE
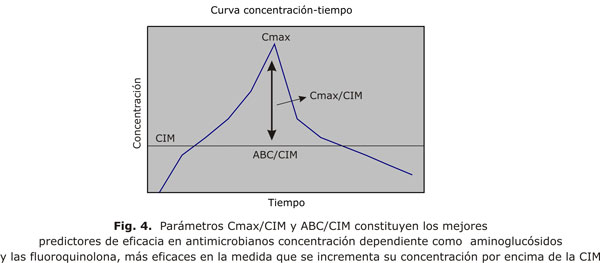
Los antimicrobianos que responden a esta cinética bactericida se caracterizan por ser más eficientes en su actividad a medida que se incrementa su concentración en el sitio de infección, generalmente son óptimos cuando su concentración supera 10 veces la CIM para el germen en cuestión; ejemplos de antimicrobianos con esta dinámica de respuesta son los aminoglucósidos24-26 y las fluoroquinolonas.22,24-27 El parámetro Cmax/CIM (fig. 4), es el mejor predictor de eficacia para este tipo de agente, además in vitro se ha comprobado que es eficaz para predecir el desarrollo de resistencia bacteriana. ABC/CIM también predice eficacia (son co-variables), ABC/CIM (fig. 4), es una medida de la exposición total del germen al antimicrobiano; Cmax/CIM y ABC/CIM son parámetros difíciles de independizar en diseños de ensayos clínicos porque cuando la Cmax/CIM es elevada, usualmente también la ABC/CIM es alta, ambos parámetros son de valor estadístico predictivo e indistinguible desde el punto de vista de cual es de importancia primaria.
{kind=link}
AGENTES QUE DESTRUYEN DE MANERA TIEMPO DEPENDIENTE (CONCENTRACIÓN INDEPENDIENTE)
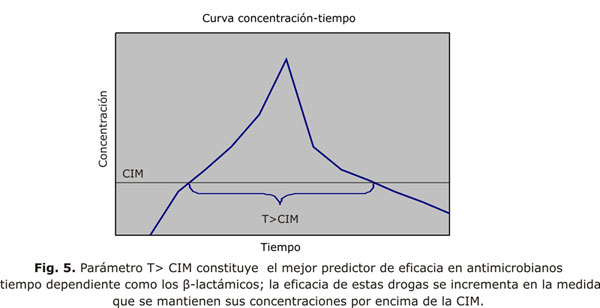
Para estos agentes en general cuando la concentración en el sitio de la infección supera 4 veces la CIM, permiten un nivel de destrucción bacteriana que no se incrementa con concentraciones superiores; el tiempo que se logra concentraciones superiores a la CIM es el que mejor predice la eficacia en esta clase de antimicrobianos; T> CIM, es un parámetro farmacodinámico que indica el periodo de tiempo en que la concentración de un antimicrobiano supera la CIM del germen en cuestión y se da en porcentaje del intervalo de la dosis (fig. 5); existen evidencias que muestran a T> CIM como un predictor farmacodinámico importante en penicilinas, cefalosporinas, carbapenémicos y monobactámicos.24,28-30 La meta en este grupo de medicamento sería prolongar la exposición de la droga. Existe diferencias entre los microorganismos en cuanto al tiempo por encima de la CIM que necesitan para ser adecuadamente eliminados; por ejemplo. penicilinas y cefalosporinas requieren menos tiempo por encima de la CIM para ser eficaces contra estafilococo que contra bacilos gramnegativos y estreptococos. Esto parece deberse a que estafilococo es el único contra el cual estas drogas exhiben "prolongado" EPA. P aeruginosa requiere un tiempo ligeramente mayor por encima de CIM que otros gramnegativo (5 al 10 % del intervalo de la dosis). Para los carbapenémicos existen mínimas diferencias en cuanto a tiempo por encima del CIM requerido para la eficacia contra la mayoría de los bacilos gram-negativos; la mayor diferencia la exhibe el S. pneumoniae con un muy corto tiempo de 10 a un 20 % del intervalo de la dosis para ser eficaz.31-33
{kind=link}
El tercer grupo destruye de manera tiempo dependiente pero difiere del grupo anterior en que posee EPA moderado o prolongado; incluye macrólidos, clindamicina, estreptograminas, tetraciclinas, oxazolidinonas, y glicopéptidos.13,22,34,35 La presencia de EPA hace que el tiempo de exposición sea menos importante, la meta serÍa proveer la cantidad adecuada de la droga. El parámetro que mejor predice la eficacia de la droga es ABC/CIM.
IMPORTANCIA CLÍNICA DE ÍNDICES Y PARÁMETROS FARMACODINÁMICOS
Conociendo que la eficacia del antimicrobiano depende del tiempo de exposición, se puede optimizar su eficacia administrándolo de manera que su concentración se mantenga lo más estable posible superando la CIM; se puede esto lograr si se utiliza las siguientes estrategias: 1) administración en infusión continua; 2) intervalos ínter dosis más cortos, aunque con concentraciones más bajas; o 3) sustituyéndolo por un antimicrobiano de mayor T1/2. El objetivo final es lograr porcentajes elevados de T> CIM.
En antimicrobianos como los aminoglucósidos en los que la eficacia depende de la concentración, independientemente del tiempo que se supere la CIM, será necesario administrar dosis más elevadas, aunque en intervalos ínter dosis más largos; de este modo se optimiza su utilización y el objetivo es lograr altos Cmáx/CIM y ABC/CIM, parámetros predictores de éxito en este grupo de medicamentos.
Por otro lado, los antimicrobianos con un EPA prolongado podrían ser administrados en intervalos aun más largos sin temor a perder eficacia, ya que los microorganismos no recrecerían durante un lapso de tiempo aunque los niveles fueran muy bajos.
Todos estos valores de predicción de actividad y parámetros asociados, como el EPA, CPM tiene una aplicación clínica muy bien definida: el diseño y optimización de las pautas de dosificación de los antimicrobianos y a largo plazo prevenir la aparición de resistencia.
ASPECTOS QUE SE DEBEN CONSIDERAR EN LA VALORACIÓN DE PARÁMETROS FARMACOCINÉTICOS/FARMACODINÁMICOS
Actualmente los parámetros farmacocinéticos/farmacodinámicos para agentes antimicrobianos se sustentan en la concentración plasmática y la CIM in vitro; el uso de la concentración total plasmática y los valores in vitrode CIM no es lo ideal. El tejido no es un compartimiento homogéneo, la distribución de la droga en plasma y tejido depende de sus características físico-químicas, el tiempo de equilibrio tiene un rango desde minutos hasta días. Implica el área de superficie capilar, el volumen de fluido en el compartimiento místico y la afectación de la distribución por las barreras anatómicas barrera hemato/encefálica (BHE), ojos, próstata, hueso; algunos antimicrobianos se unen de manera importante a proteínas hísticas o celulares. Es común creer que la concentración del antibiótico libre en equilibrio de concentración en plasma y en fluido hístico es igual, basados en el precepto de que la fuerza de distribución es la difusión pasiva; de este modo se trata de predecir la concentración hística infiriéndola de la plasmática, pero esto no siempre es correcto; la diferencia entre la concentración total del plasma y la concentración libre en tejido puede ser significativa en innumerables situaciones, como en drogas de alta unión a proteínas, y la penetración en los tejidos de difícil acceso. Todo esto hace que la concentración plasmática total no sea un valor de salida ideal farmacocinético para definir dosis de medicamento. La penetración a tejidos depende de variables como difusión, transporte activo, liposolubilidad, y unión a proteínas; por ejemplo, en infecciones del sistema nervioso central, los antimicrobianos lipofílicos no ionizados como rifampicina y metronidazol penetran ampliamente, mientras que la mayoría de los beta-lactámicos, quinolonas y glicopéptidos tiene una penetración limitada y requieren ser administrados en dosis máximas que se puede ver favorecida por el aumento de permeabilidad que acompaña a la infección, los aminoglucósidos y las cefalosporinas de 1ª y 2ª generación tienen mínima penetración. En osteomielitis la penetración del antimicrobiano también es clave para el éxito de la terapia, las lincosaminas tienen alta penetración; vancomicina y quinolonas logran concentraciones superiores a la CIM de los principales patógenos. Solo la concentración de la droga libre en el sitio diana es responsable del efecto terapéutico, por lo que el parámetro farmacocinético ideal para el análisis farmacocinético/farmacodinamico sería la concentración de la droga libre en el fluido intersticial.
LA APLICACIÓN DE LOS PRINCIPIOS FARMACODINÁMICOS AL PACIENTE CRÍTICO
La aplicación de los principios farmacodinámicos en el paciente grave es complicado por los cambios farmacocinéticos que se operan en estos pacientes, el incremento en el Vd, decrecimiento en las concentraciones de proteínas séricas, decrecimiento en el metabolismo y aclaramiento de la droga (disfunción de órgano o hipo perfusión), incremento del metabolismo y aclaramiento por el estado hipermetabólico del paciente crítico.36 A pesar de los cambios inherentes al paciente crítico, la optimización de las dosis basadas en la caracterización farmacocinética de este y la apropiada aplicación de los principios farmacodinámicos ofrecen un potencial indiscutible para mejorar la sobrevida del paciente y para prevenir la emergencia de resistencia. Por la severidad de la infección en el paciente crítico y las variabilidades farmacocinéticas, así como las relacionadas con la penetración en tejido, la recomendación general para la dosis de antimicrobianos son estrategias de dosis agresivas; bajas dosis de antimicrobianos pueden fallar para erradicar el patógeno y predisponen al desarrollo de resistencia. El uso de altas dosis de medicamento potencialmente compensan las alteraciones farmacocinéticas presentes, lo que incrementa la posibilidad de alcanzar las metas farmacodinámicas adecuadas que se correlacionan con éxito terapéutico, pero no se debe perder la perspectiva del incremento en las reacciones
adversas.
adversas.
La farmacodinamia permite al clínico seleccionar la droga más potente y ofrece una guía para definir la dosis e intervalos de dosis más inocuos y eficaces para un patógeno particular en un sitio específico de infección. Para la industria farmacéutica la aplicación de estos principios ayuda a predecir las probabilidades de éxito del compuesto en desarrollo y guía a la hora de definir regímenes de dosis en el diseño de ensayos clínicos. Dado que la introducción de nuevos agentes antimicrobianos para patógenos multi-droga-resistentes puede tomar décadas para estar disponibles en el arsenal terapéutico, la opción más inmediata es optimizar el uso de los que se tienen a nuestra disposición.
No hay comentarios:
Publicar un comentario