EL SUELO: CONCEPTO Y FORMACION
Alteración química
En contacto con el aire, y sobre todo con el agua, los minerales de las rocas se alteran. Por otra parte, los organismos atacan a los minerales para extraer elementos nutrientes (K, Ca, Mg...) y transforman a los minerales.
La alteración química del material original, se encuentra ampliamente desarrollada en los suelos y se puede poner de manifiesto simplemente comparando la mineralogía inicial de la roca frente a la mineralogía del suelo que se forma a partir de ella.
| cuarzo | ortosa | albita | biotita | moscovita | piroxeno | ilita | caolinita | ||
| Suelo | Hor. A | 62% | 14% | 3% | 1% | 5% | 0% | 10% | 5% |
| Hor. B | 55% | 18% | 6% | 5% | 6% | 1% | 6% | 3% | |
| Hor.C | 52% | 20% | 8% | 10% | 7% | 3% | 0% | 0% | |
| Roca | Hor. R | 48% | 22% | 8% | 12% | 7% | 3% | 0% | 0% |
También se puede evaluar el grado y el tipo de alteración sin más que hacer un estudio de cualquier muestra de suelo en el microscopio petrográfico.

Los principales procesos de alteración química son:
NaCl + H2O <==> Cl- + Na+ + H2O
halita
CaSO4 + 2H2O <==> CaSO4.2H2O
anhidrita yeso
CaAl2Si2O8 + 8H+ <==> Ca++ + 2Al3+ + 2H4SiO4
feldespato (anortita) ac. metasilíco
Fe(OH)3 + 3H+ e- <==> Fe++ + 3H2O
Lo que acabamos de exponer se refiere fundamentalmente a la fracción mineral, pero el material orgánico también sufre una intensa transformación.
En el caso concreto de la materia orgánica la alteración puede conducir al desarrollo de dos procesos distintos: humificación y mineralización. Ambas inicialmente tienen una misma vía de actuación, la transformación de los restos vegetales y animales al morir, pero desembocan en dos resultados completamente distintos. La humificación engloba a una serie de procesos de alteración entre productos orgánicos, es decir que siempre se conserva la estructura orgánica. Por tanto la humificación conserva el material orgánico en el suelo, forma el humus. Por el contrario la mineralización conduce a la destrucción total de los restos orgánicos descomponiéndolos en sus productos inorgánicos sencillos (H2O, CO2, NH3...) eliminándose (realmente mineralizándose) gran parte de la materia orgánica del suelo.
4.2.3 Translocación
Además de estos dos procesos de desagregación física y alteración química hay un tercer proceso que ejerce una importantísima acción en la formación del suelo y es la translocación de sustancias, que por un lado mezcla y agrega los materiales edáficos y por otro lado, los separa y los concentra. Todas estas acciones se realizan bien por los organismos del suelo, muy especialmente por los que excavan galerías, como las lombrices y las hormigas o por simple efecto mecánico, muy frecuentemente por la acción del agua que transporta los materiales, a veces en suspensión a veces en disolución. Este arrastre por la acción del agua ejerce efectos muy importantes en el suelo y puede eliminar a las sustancias transportadas fuera del perfil o acumularlas a una determinada profundidad.
La translocación de sustancias también se puede demostrar fácilmente viendo por ejemplo sustancias que tapizan las paredes de los poros e incluso rellenando completamente las grietas del suelo o simplemente observando el material que rellena las galerías de la fauna o también por los montoncitos acumulados en las entradas de los hormigueros y toperas.

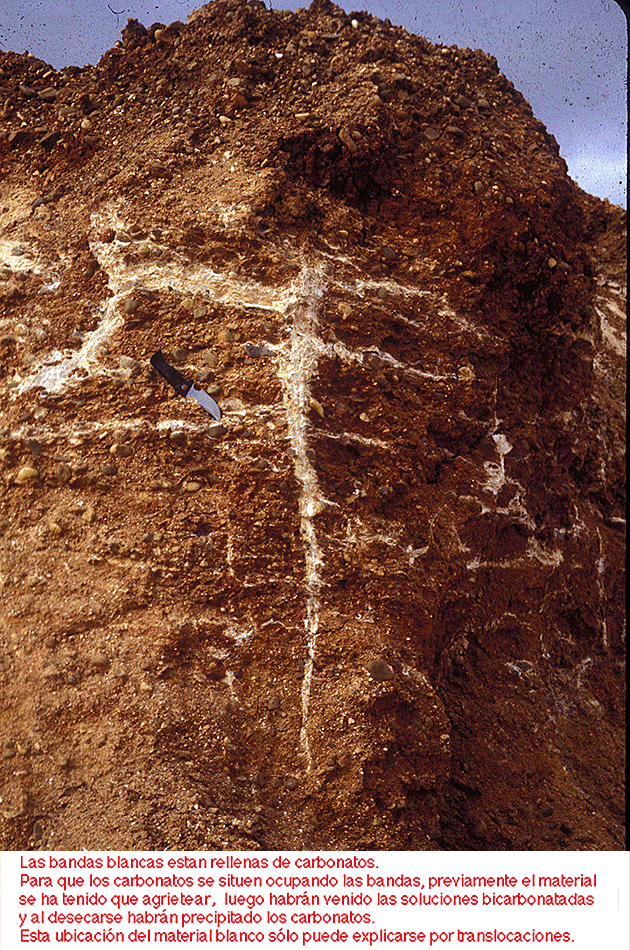
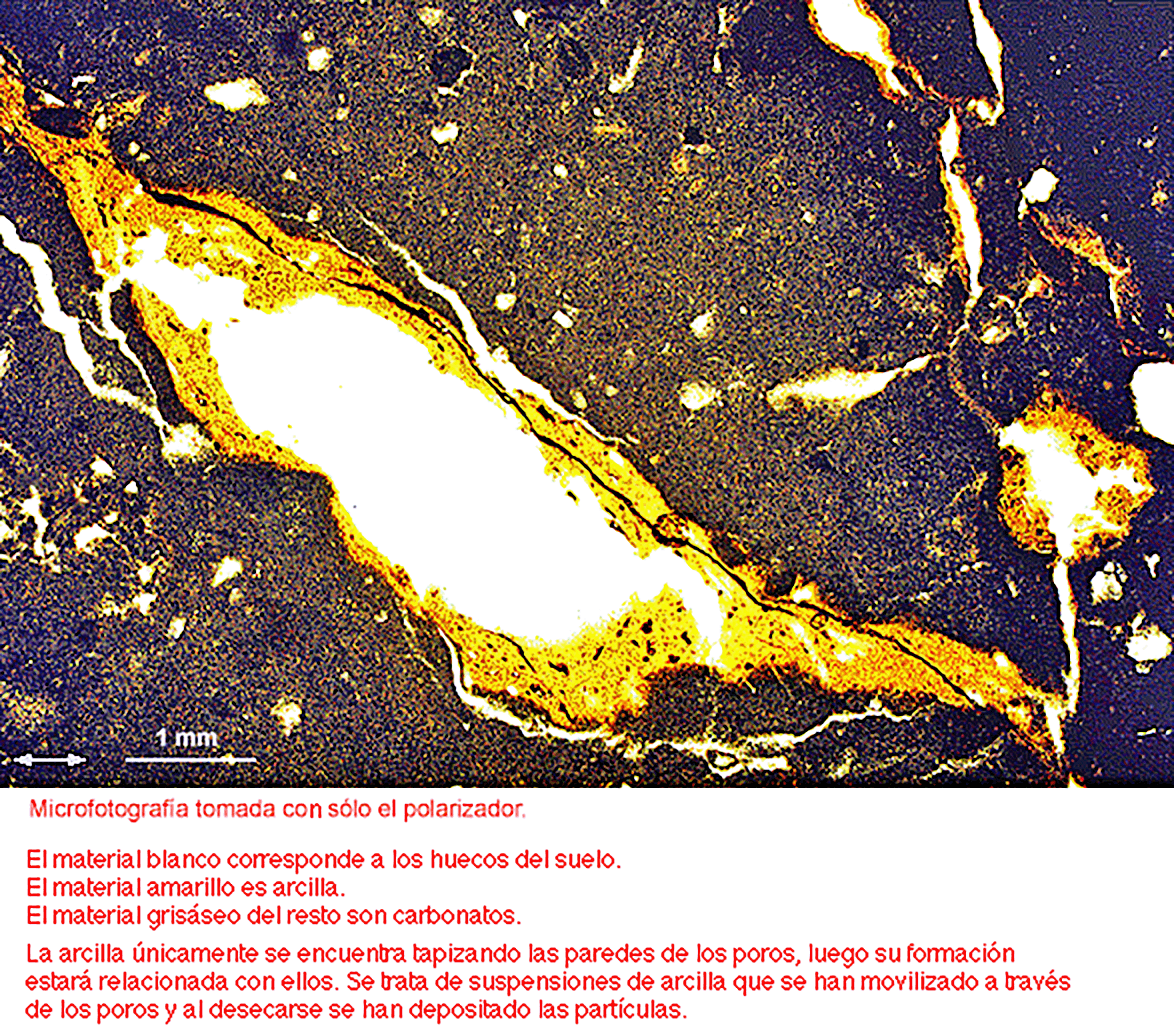
Es decir que el proceso de translocación de materiales en el suelo es muy complejo afectando a muy distintas sustancias (minerales, materia orgánica y complejos organo minerales, ya sean como soluciones o suspensiones) y por muy diferentes causas (gravedad, capilaridad, evaporación, actividad biótica, o como consecuencia del hinchamiento y contracción de la masa del suelo).
Los procesos de deterioro de tipo químico pueden definirse como aquellos que implican un cambio en la composición química global y/o mineralógica de la roca original. Generalmente, estos procesos de alteración suponen la interacción de una solución acuosa (y/o gases) con el material rocoso, produciendo una solución de composición diferente de la inicial y un residuo de sólidos insolubles (fases minerales secundarias, esto es, formadas en el proceso de alteración). El medio de alteración transporta parte de los productos fuera del sistema rocoso, cambiando la composición original del mismo.
Como para cualquier reacción química, los procesos de alteración químicos están controlados por variables fisico-químicas (composición química y mineralógica de las rocas, composición de las disoluciones acuosas y composición de los gases, temperatura, pH, Eh,...). Es por tanto necesario un conocimiento preciso de estas variables a la hora de describir los procesos de deterioro.
La composición de la roca original (e indirectamente sus características texturales y estructurales) es un control decisivo sobre que tipo de proceso será operativo y sobre el resultado del mismo. Por lo tanto, las distintas reacciones químicas se describen a continuación en relación con los tipos de rocas sobre los que apliquen de manera específica.
2.EQUILIBRIO QUIMICO
Los átomos que forman cualquier sistema fisico-químico tienden a ordenarse en fases (minerales, soluciones líquidas y gaseosas) cuya composición y estructura dependen de las condiciones ambientales bajo las que está sometido el sistema. Cualquier ordenamiento particular de un sistema concreto en determinadas asociaciones de fases puede permanecer estable o sufrir cambios hacia otro tipo de ordenamiento distinto. La situación estable se denomina de equilibrio fisico-químico o termodinámico. Por ejemplo, en un sistema en que coexistan agua y una sal bajo unas condiciones ambientales determinadas, la condición de equilibrio se adquiere al disolverse una cantidad definida de sal en el agua. Esta situación se mantendrá si las condiciones ambientales no cambian, lo cual no implica que el sistema se comporte estáticamente, esto es, que no exista movilidad de los átomos. De hecho, la condición de equilibrio en un sistema se describe en términos de ecuaciones químicas, de manera que los cambios que se producen en una dirección de la reacción son compensados con los producidos en el sentido contrario. En el ejemplo anterior, una vez alcanzado este estado de equilibrio se producirá un proceso de disolución de la sal que será compensado por la precipitación de la misma cantidad de sal a partir de la disolución, de manera que el sistema permanece sin cambios globales en las cantidades y composiciones de la sal y la disolución. Esta condición de equilibrio se describe definiendo las condiciones ambientales y las proporciones y concentraciones de las fases.
El cambio de una o más de las variables implica una tendencia al cambio en el ordenamiento de la materia. En el ejemplo anterior, se producirá un desplazamiento de la reacción hacia la disolución o precipitación, según el caso. El nuevo ordenamiento nace de la condición de desequilibrio del sistema para las nuevas condiciones, y se detendrá cuando se alcance un ordenamiento nuevo en equilibrio para estas condiciones. Así, para que un sistema alcance el equilibrio debe mantenerse sometido a una condiciones constantes durante cierto tiempo, ya que las velocidades de las reacciones que afectan al sistema no son muy elevadas. Si las condiciones cambian a más velocidad que las velocidades de reacción, no dará tiempo a reorganizar la materia. Por lo tanto, es importante tener en cuenta que las ecuaciones químicas indican los cursos posibles de las reacciones químicas, pero no informan de las velocidades de las mismas que son las que realmente determinan si la reacción es importante o no en los procesos de alteración. Estos aspectos son tratados por la cinética de reacción.
Cualquier desviación de las condiciones de equilibrio en un sistema tiene como resultado la transformación del sistema mediante uno o varios procesos fisico-químicos que tienden a reorganizar la materia y restablecer el equilibrio. Esta reorganización se realiza mediante cambios en la composición de las fases existentes, produciendo fases nuevas, y/o destruyendo fases anteriores. Los procesos de deterioro que afectan a los materiales pétreos utilizados como materiales de construcción y ornamentación resultan de la condición de desequilibrio a que están sometidas a las asociaciones de fases que constituyen las rocas bajo las condiciones del medio ambiente atmosférico. No obstante, los procesos de alteración rara vez consiguen alcanzar el equilibrio en el medio ambiente atmosférico, ya que el sistema no permanece cerrado químicamente y la velocidad de cambio de las condiciones ambientales es mayor que las velocidades de reacción.
En consecuencia, los factores cinético son muy importantes en la evaluación de los procesos químicos. Su importancia queda manifestada en el hecho de que la mayoría de los minerales que forman las rocas no son estables en las condiciones superficiales, permaneciendo el ellas metaestablemente debido a las bajas velocidades de reacción a baja temperatura. Aunque las bajas temperaturas a que se verifican los reacciones de alteración condicionan bajas velocidades de reacción, este efecto se ve compensado por la presencia de fases fluidas (soluciones acuosas y/o gaseosas) en la mayoría de las reacciones. Esto último aumenta las velocidades de reacción ya que las velocidades de difusión de las especies químicas incrementan de manera importante en presencia de fases fluidas que sirven de transporte. Por ello, la presencia de fases fluidas es de crucial importancia en el proceso de alteración de un material al servir de medio de transporte de los productos de alteración. Así, si estos son retirados del sistema las reacciones progresarán en la misma dirección, mientras que si permanecen en el mismo, el sistema puede devenir cerrado y por lo tanto alcanzar el estado de equilibrio, llegándose a parar la reacción.
Resumiendo, podemos establecer como características generales de los procesos de alteración en el medio ambiente atmosférico las siguientes:
· Condiciones de desequilibrio en el material original, lo cual supone fuerte tendencia al cambio de ordenación de la materia.
· Condiciones ambientales muy cambiantes, lo que supone ausencia de equilibrio.
· Presencia de fases fluidas que interaccionan con el material, lo que supone velocidades de reacción más elevadas que en su ausencia y capacidad de transporte fuera del sistema de los productos de reacción.
· El sistema será por lo general abierto y no alcanzará el equilibrio, por lo que las reacciones procederán en el mismo sentido, esto es, en el sentido de la alteración. El resultado es que será imposible detener el proceso de deterioro.
A todo esto hay que añadir la acción de la contaminación y de ciertos organismos, que en general suponen el aceleramiento de los procesos de alteración. Aunque los efectos de la contaminación pueden tratarse al mismo tiempo que los procesos de tipo químico, serán considerados aparte, al igual que los efectos debidos a la acción organismos.
De los tipos de reacciones químicas operativas en los procesos de alteración, la disolución es muy importante ya que los productos de reacción suelen ser transportados fuera del sistema. Laoxidación se producirá en los minerales que presenten componentes químicos con estados de oxidación variables al ponerse en contacto con el oxígeno atmosférico. Procesos de reducción son muy raros en el ambiente superficial rico en oxígeno libre, aunque pueden ser operativos en relación con ciertos procesos de origen orgánico. La hidrólisis es importante el los procesos de alteración de silicatos. Otros tipos de reacciones son la carbonatación, hidratación y quelación, aunque este último en relación con procesos biológicos y de contaminación.
3.FACTORES FISICO-QUIMICOS
3.1.PRESENCIA DE AGUA
Los procesos de alteración que vamos a considerar son esencialmente reacciones que tienen lugar en presencia de agua. El agua es un líquido poco usual. Es un disolvente excelente, tanto por la variedad de sustancias que puede disolver como por la cantidad de las mismas. La estructura de su molécula (H2O) es la clave de este comportamiento. Los dos átomos de hidrógeno están embebidos dentro del átomo de oxígeno, por lo que la molécula es casi esférica y presenta un radio poco mayor que el radio del oxígeno. El ángulo entre las líneas que unen los centros de los átomos de hidrógeno con el centro del átomo de oxígeno es de 105o. Esta estructura resulta en una molécula con distribución preferente de cargas. En y entre los átomos de hidrógeno se concentra la carga positiva, mientras que la negativa lo hace en el polo opuesto. Por lo tanto, la molécula de agua es un dipolo. Las atracciones electrostáticas de estos dipolos hacen que las fuerzas cohesivas entre las moléculas de H2O sean mucho más fuertes que las de otros líquidos. Por esto, para ser una sustancia de bajo peso molecular, el agua presenta valores altos de puntos de fusión y ebullición. Además, sus buenas cualidades como disolvente proceden de esta misma causa, ya que los iones son atraídos por los polos de carga opuesto de las moléculas de agua, permitiendo su dispersión en el líquido.
3.2.POTENCIAL IÓNICO
Como acabamos de ver, los iones en solución atraen las moléculas de agua; los cationes atraen los polos negativos y los aniones los positivos. El número de moléculas de agua que puede atraer un ión determinado será función de su radio y su carga. A mayor radio y carga, mayor número de moléculas atraídas. Sin embargo, el factor esencial es la intensidad de las cargas eléctricas en la superficie externa de los iones, que puede describirse como el potencial iónico. Este valor es la relación entre la carga de un ión (Z) y su radio (r). Por lo tanto, la tendencia a la hidratación de un ión en solución y su comportamiento puede medirse por su potencial iónico (Figura 1):
· Los cationes con potencial iónico bajo (menor de 3), tales como los alcalinos (Na+, K+) y alcalinotérreos (Ca2+, Mg2+), atraen con intensidad moderada a las moléculas de agua, permaneciendo en solución durante los procesos de alteración.
· Los cationes con potencial iónico intermedio (3-12), tales como Al3+, Fe3+ y Si4+, atraen con mayor fuerza las moléculas de agua, pero al mismo tiempo repelen con mayor fuerza los átomos de hidrógeno de las mismas, pudiendo forzar el desprendimiento de uno de ellos. Se forma así una molécula de (OH)–, cargada negativamente, que se combina con el catión para formar un hidróxido insoluble que precipita (e.g., Al(OH)3, Fe(OH)3). Por esta razón, este tipo de cationes suelen formar precipitados por hidrólisis, que son muy poco solubles.
· Los cationes con potenciales iónicos más elevados (más de 12), tales como C+4, N+5 y S+6, repelen con tal fuerza a los átomos de hidrógeno de las moléculas de agua, que todos ellos son desprendidos de las mismas. Los iones de oxígeno libres se combinan con los cationes, formándose moléculas de oxiácidos (e.g. CO3=, NO3–, SO4=). Por esta razón estos elementos son de nuevo solubles bajo la forma de aniones complejos.

Figura 1. Separación de los cationes en términos de potencial iónico (carga/radio).
3.3.CONCENTRACIÓN DE IONES HIDRÓGENO (pH)
La concentración de iones hidrógeno del agua es de gran importancia en las reacciones de alteración. El ión hidrógeno (H+) está siempre presente en cualquier disolución acuosa. Esto se debe a que el agua tiende a disociarse en cierta proporción según la reacción:
H2O = (OH)– + H+ (1)
que tiene una constante de ionización (a 25 oC) de:
K = [H+]*[OH–]/[H2O] = 1.82*10-16 (2)
Puesto que la concentración de las moléculas de agua es muy alta comparada con las concentraciones de los iones hidrógeno e hidroxilos, la constante de ionización o de disociación suele expresarse exclusivamente en términos del producto de las concentraciones de los iones hidrógeno e hidroxilos. En este caso, la constante se denomina constante del producto iónico del agua, esto es (a 25 oC):
Kw = [H+]*[OH–] = 1.008*10-14 » 10-14 (3)
[H+] = [OH–]/Kw (4)
Estas relaciones no son constantes, sino que dependen de las sustancias disueltas en el agua (ácidos o bases) y de la temperatura. Así, el agua se denomina neutra cuando presenta [H+] = [OH–] (= 10-7 a 25 oC), agua ácida cuando [H+] > [OH–] (>10-7 a 25 oC), y agua básica cuando [H+] < [OH–] (<10 span="">-7 a 25 oC). Para expresar las variaciones posibles en las concentraciones de los iones hidrógeno e hidroxilo (que varían de 1 a 100 billones) se establece el potencial de hidrógeno o pH, que se define como:
pH = log[H+] (5)
de manera que una disolución neutra presenta un pH igual a 7, una ácida de 0 a 7, y una básica de 7 a 14 (a 25 oC).
La solubilidad de muchas sustancias están fuertemente influenciadas por el pH. Especialmente importante es en el caso de la precipitación de hidróxidos. Así por ejemplo, la solubilidad del hierro es 100000 veces mayor a pH = 6 que a pH = 8. Por lo tanto, el hierro puede disolverse en soluciones débilmente ácidas y precipitar como hidróxido de Fe3+ si la solución se convierte en básica. Las solubilidades del Al y Si también están fuertemente condicionadas por el pH (Figura 2), lo cual influye en el tipo de alteración en rocas silicatadas. En condiciones muy ácidas (pH<4 5="" 9="" al.="" al="" aluminio="" arcillas="" aumenta="" aumentar="" bajo="" cada="" condiciones="" de="" dej="" del="" el="" en="" enriquecido="" entre="" es="" estas="" form="" fuera="" geles="" hacerse="" hidr="" insoluble="" la="" las="" lice.="" lo="" m="" minerales="" ndolo="" ndose="" o="" pero="" ph.="" ph="" puede="" que="" residuo="" rico="" rocoso="" s="" ser="" si="" silicio="" sistema="" solubilidad="" soluble="" span="" supone="" transportado="" vez="" virtualmente="" xidos="" y="">
En la Figura 3 pueden observarse las condiciones de pH de algunos tipos de aguas naturales. Las condiciones apropiadas para la alteración de materiales pétreos en el ambiente atmosférico está señalada por una línea, que hacia la parte inferior del diagrama se señala discontinua para indicar condiciones poco probables. No obstante, hay que tener en cuenta que la contaminación atmosférica puede distorsionar este campo, pudiendo alcanzarse valores de pH bastante bajos (menores de 4).

Figura 2. Solubilidad de sílice y alúmina en función del pH (Tomado de Mason, 1966).
3.4.POTENCIAL DE OXIDACIÓN-REDUCCIÓN (Eh)
Muchos elementos se presentan en estados de oxidación variables. El más común y abundante es el hierro, que se presenta como metal nativo (Fe), compuestos de ferrosos (Fe2+) y férricos (Fe3+). Otros elementos son Mn (2,3,4), S (-2,0,6), Cu (0,1,2), Co (2,3), etc.
La estabilidad de un elemento en un estado de oxidación determinado depende del incremento de energía involucrado en las pérdidas o ganancias de electrones. Una medida cuantitativa de este cambio de energía lo aporta el factor llamado potencial de oxidación-reducción o potencial redox, simbolizado como Eo o Eh.
El valor del potencial de oxidación de cualquier reacción redox se establece en una escala relativa respecto de la energía involucrada en la reacción:
H2 = 2H+ + 2e– (6)
Esto es, la energía necesaria para liberar dos electrones de una molécula de hidrógeno gaseoso es tomada como estándar de comparación, por lo que el valor del potencial redox de esta reacción se fija en 0.00 eV, extendiéndose la escala de Eh a valores positivos y negativos. A medida que el valor de Eh se hace menor, la energía necesaria para desplazar la reacción hacia la pérdida de electrones (esto es, en el sentido de la oxidación) se hace menor. Este tipo de reacciones no se producen aisladamente, sino que están acopladas en pares, de manera que al oxidarse un compuesto, se reduce otro. Por ejemplo:
4(OH)– = O2 + 2H2O + 4e– (Eh = 0.40) (7)
Fe(OH)2 + (OH)– = Fe(OH)3 + e– (Eh = -0.56) (8)
restando 4(8) - (7):
4Fe(OH)2 + O2 + 2H2O = 4Fe(OH)3 (9)
de manera que el hierro se oxida, perdiendo 4 e– que son tomados por las dos moléculas de O2, que se reducen. El oxígeno, por lo tanto, es oxidante en esta reacción.
Cualquier otro par de ecuaciones se pueden acoplar en los procesos de oxidación-reducción. En estas reacciones, la forma reducida de cualquier ecuación tiene la suficiente energía para reducir la forma oxidada de otra ecuación con potencial redox mayor.
Los valores de potenciales de oxidación varían con la concentración de las sustancias reactantes. Estas variaciones con la concentración son de especial importancia en las reacciones que involucran iones hidrógeno o hidroxilos. Por lo tanto, las variaciones en el pH, indicativas de variaciones en las concentraciones de H+ y (OH)–, producen cambios en los valores de Eh de las reacciones en consideración. En general, el incremento del pH (o sea, a medida que la concentración de H+ desciende) para una misma reacción supone una disminución de los valores de Eh. Además, para condiciones variables de pH pueden producirse inversiones en las capacidades relativas de reducción-oxidación de un par de reacciones concretas.
Las condiciones de Eh del medio ambiente determinan las reacciones que pueden tener lugar. Así, en el medio ambiente atmosférico, las reacciones que pueden tener lugar están teóricamente limitadas a aquellas cuyos valores de Eh estén comprendidos entre 1.23 y 0.00 eV, los valores de Eh de las reacciones de descomposición del agua (Figura 3):
2H2O = O2 + 4H+ + 4e– (Eh = 1.23) (10)
2H+ +2e– = H2 (Eh = 0.00) (6)
Los valores de Eh de estas dos ecuaciones son para valores de pH = 0 (esto es, concentración de H+ = 1) y 25 oC de temperatura. El cambio del Eh en función del pH, a 25 oC, es de 0.06 eV por cada unidad de pH. La mayoría de las aguas que interaccionan en el medio ambiente presentan valores de pH entre 4 y 10. Para unas condiciones neutras (pH = 7), los valores de Eh de las reacciones (10) y (6) son 0.82 y -0.41 eV respectivamente, lo que indicaría que los potenciales redox del medio ambiente en esas condiciones de pH y temperatura (25 oC) deberían estar entre esos márgenes. Sin embargo, el hecho de que los procesos de alteración de los materiales pétreos utilizados como materiales de construcción se produzcan en el ambiente atmosférico, esto es, con oxígeno libre, asegura unas condiciones de Eh oxidantes independientemente de los valores de pH, y por lo tanto más próximas a los valores de la reacción (10) para cada valor de pH.
Los procesos de disolución y transporte de elementos que presentan varios estados de oxidación están fuertemente controlados por los potenciales de oxidación del medio. Como hemos visto, los valores de los potenciales redox de muchas reacciones de oxidación descienden al aumentar el pH, lo cual supone que a medida que las soluciones son más alcalinas (pH>7) la oxidación será más rápida.

Figura 3. Aguas naturales en relación con Eh y pH (tomado de Henderson, 1982).
4.TIPOS DE REACCIONES QUIMICAS
A continuación se describen brevemente los tipos más importantes de reacciones químicas que afectan a los materiales pétreos. La mayoría de estas reacciones incluyen al agua como fase activa, produciéndose iones que entran en solución, por lo que estas reacciones pueden considerarse globalmente como reacciones de disolución. Por lo tanto se analizarán las reacciones de disolución globalmente, y subsecuentemente se subdividirán en tipos específicos.
La disolución, entendida como ataque químico del agua, es el estadio principal de la alteración química. Puede tener lugar en superficies expuestas al agua de lluvia, o en una delgada película de agua adsorbida alrededor de las partículas sólidas en el interior del material. La cantidad de disolución o alteración depende de la solubilidad del sólido y de la cantidad de agua que atraviesa la superficie expuesta a la disolución.
Los minerales pueden disolverse en el reactante acuoso vía dos tipos de reacciones:
Reacciones de disolución congruente, en las que el mineral es disociado y entra en disolución en forma de iones simples, como por ejemplo:
ClNa + H2O = Cl– + Na+ + H2O (11)
o en forma de iones complejos generados por reacción con el agua, como por ejemplo:
CaCO3 + H2O = (HCO3)– + Ca2+ + OH– (12)
En este tipo de reacciones el sólido entra en la disolución directamente, sin formar otras fases producto de reacción, siendo el caso de las sales solubles, de algunos silicatos simples y de la calcita.
Reacciones de disolución incongruente, en las que el mineral reacciona con el agua, descomponiéndose en una fase sólida distinta de la reactante y en especies químicas que entran en solución, como por ejemplo:
MgCO3 + 2H2O = Mg(OH)2 + (HCO3)– + H+ (13)
Este segundo tipo es el caso de silicatos complejos y algunos carbonatos.
Cuando el mineral se disuelve vía reacciones de disolución congruentes en forma de iones simples el proceso de denominan simplemente de disolución. En el caso de que el mineral entre en solución vía reacciones congruentes o incongruentes en forma de iones complejos formados por reacción con el agua, como serían los casos de los silicatos y algunos carbonatos, el proceso se denomina de hidrólisis. En realidad todas las reacciones de disolución son reacciones de hidrólisis, ya que el agua reacciona en mayor o menor medida con el sólido, variando las condiciones de pH de la solución. No obstante, suele reservarse el concepto de hidrólisis para las reacciones donde el agua reacciona con el catión de la sal para producir un compuesto generalmente poco soluble.
Un caso particular de disolución congruente es la disolución de carbonatos (esencialmente calcita), donde el agua y CO2 intervienen para formar iones complejos que reaccionan con el carbonato. Dada su importancia, este tipo de procesos se consideran a parte como disolución de carbonatos.
Otro caso particular de disolución es la carbonatación de silicatos, donde el agua y CO2 de nuevo intervienen para formar iones complejos que atacan los silicatos formándose ácidos de C de manera similar a la disolución de carbonatos. Este tipo de ataque a silicatos puede ser significativo en ambientes ácidos contaminados, denominándose carbonatación.
Un tipo de reacciones donde generalmente entra el agua como agente activo, aunque a veces puede no intervenir, son las reacciones de oxidación-reducción. Estas reacciones afectan a los minerales que contienen elementos con estados de oxidación variables, aunque el más importante es el Fe (Fe+2 y Fe+3). Así, estos minerales con Fe2+, como sulfuros de Fe (pirita), óxidos de Fe (magnetita), silicatos de Fe (piroxenos, olivino) y material artificial variado donde el Fe2+ sea un componente esencial, presentarán estos procesos de óxidación.
Otro tipo de reacciones particulares son las reacciones de quelación, donde entran en reacción el agua y el CO2. Este tipo de reacciones químicas están relacionado con la actividad orgánica de ciertas plantas y líquenes, por lo que se tratan en otro capítulo.
4.1.DISOLUCIÓN DE SALES
Como hemos visto, la disolución la consideramos como el proceso de disociación de un compuesto en sus iones simples constituyentes. Este tipo de procesos se da esencialmente en las sales solubles via reacciones de disociación tales como (Sólidos = iones libres):
NaCl = Na+ + Cl– (11)
CaSO4 = Ca2+ + (SO4)= (14)
CaCO3 = Ca2+ + (CO3)= (15)
Generalmente la solubilidad de las sales es mayor al aumentar la temperatura, aunque en algunos casos esta tendencia puede invertirse, como en el caso del yeso cuya solubilidad máxima está en torno a 40 oC (Figura 4); en otros casos, la solubilidad desciende con la temperatura, como en el caso de la calcita (Figura 5). Por otra parte, para las mismas condiciones de P y T, los distintos compuestos tienen solubilidades distintas; así, la halita es más soluble que la anhidrita y esta última más soluble que la calcita. En la Tabla 1 se expresan las solubilidades de distintos minerales. La presencia de sales solubles en los materiales, ya sean de origen primario (morteros de yeso o mixtos, alabastro,...), o secundario, ejerce una fuerte influencia en el deterioro del material.

Figura 4. Solubilidad del yeso en agua en función de la temperatura (tomado de Winkler, 1973).
En primer lugar hay que considerar la propia disolución de las sales, que genera pérdidas importantes de materia si el medio acuoso está en contacto con la superficie atmosférica. Cuanto mayor sea la cantidad de agua que circula por el material y su velocidad, y menor sea su grado de saturación, mayor será la tasa de disolución de las sales y la pedida de materia. Esto se debe a que si el agua circula por el sistema poroso, accediendo al exterior, será poco probable alcanzar el grado de saturación, por lo que el sistema no alcanza el equilibrio y las reacciones de disociación no se interrumpen, continuando desplazadas hacia los productos de reacción. Además, si la disolución original es diluida para las condiciones ambientales bajo las que se produce la disolución, las reacciones de disolución son más rápidas. Si además consideramos altas velocidades de circulación del agua, la disolución aumenta rápidamente (Figura 6).

Figura 5. Solubilidad de la calcita en función del CO2 disuelto en la atmósfera y la temperatura (tomado de Winkler, 1973).

Figura 6. Influencia del flujo de agua en la tasa de disolución de calizas por soluciones diluidas de HCl (Tomado de Winkler, 1973).
Por lo tanto, las zonas de los materiales expuestas a cantidades importantes de disoluciones diluidas y con rápida circulación son las más propensas a la disolución, tales como paramentos expuestos al agua de lluvia, cornisas, fracturas abiertas, etc. Por esto hay que evitar la utilización de materiales salinos (esencialmente yeso) en zonas expuestas al exterior. En las partes interiores del sistema poroso, se producirán disoluciones importantes sólo en las zonas bajas, donde el ascenso capilar puede ser intenso, aunque normalmente las sales vuelven a precipitar dentro del sistema poroso o formando eflorescencias y costras externas por evaporación, por lo que el proceso de pérdida de materia por disolución es más lento. En cualquier caso, debe recordarse que las características climáticas del medio (esencialmente pluviosidad) son críticas.
En segundo lugar hay que considerar el efecto químico que la presencia de sales tiene en el sistema poroso de las rocas. Como ya se sabe, las sales son compuestos iónicos muy higroscópicos, esto es, tienen gran facilidad de adsorber agua es su superficie. Esta película de agua se satura en la sal con la que está en contacto, lo que facilita el intercambio iónico con minerales adyacentes aumentando la disolución de los mismos. Este efecto es más acusado cuanto más soluble sea la sal. Un ejemplo sería la interacción del NaCl con CaCO3. La película de agua superficial de la halita se satura en NaCl; el Na+ de la disolución puede difundirse a través de ella y entrar en contacto con la calcita, de manera que se intercambia Ca del carbonato por Na de la disolución; de esta manera se disuelve el carbonato cálcico y se forma carbonato sódico, que es muy soluble. El proceso se detiene cuando se alcanzan las condiciones de equilibrio, esto es, cuando la solución se satura en NaCl, Na2CO3 y CaCO3, aunque si la película de agua está en contacto con una solución no saturada en circulación, el sistema no alanza el equilibrio y contínua hasta la completa disolución del carbonato o la sal.
4.2.DISOLUCIÓN DE LOS CARBONATOS
Muchos carbonatos sufren meteorización química por disolución congruente a través de ataque ácido. El ácido se forma por reacción entre el dióxido de carbono (CO2) procedente de la atmósfera o de la respiración orgánica y el agua. Esta reacción es la siguiente:
CO2 + H2O = (HCO3)– + H+ (16)
La reacción de disolución congruente puede escribirse (utilizando la calcita como ejemplo):
CaCO3 + H2O = (HCO3)– + Ca2+ + OH– (12)
despejando H2O de (16) y sustituyéndolo en (12) tenemos:
CaCO3 + (HCO3)– + H+ = (HCO3)– + Ca2+ + OH– + CO2 (12’ )
que puede simplificarse en:
CaCO3 + H+ = Ca2+ + OH– + CO2 (12’’)
o finalmente en:
CaCO3 + H+ = Ca2+ + (HCO3)– (17)
De estas ecuaciones se deduce que existe una relación directa entre las cantidades de CO2 y CaCO3 disueltas en el agua, o también, que cuanto más ácida es la disolución, mayor es su capacidad de disolver CaCO3.
Si se considera un sistema en equilibrio a una presión constante (fijada en nuestro caso a 1 atm), existirá una relación entre la concentración (o presión parcial) de CO2 en la atmósfera y la cantidad de carbonato disuelto en el agua en equilibrio con la misma. Esto se debe a que la concentración de CO2 en el agua es función de su concentración en la atmósfera. En la Figura 7 se observan las relaciones entre la cantidad de CO2 en la atmósfera y las solubilidades máximas de CO2 en agua en equilibrio con la atmósfera para distintas temperaturas. Puede apreciarse como para una misma cantidad de CO2 en la atmósfera, la cantidad disuelta en agua disminuye con la temperatura, y que, lógicamente, la cantidad de CO2 disuelta en el agua aumenta con la cantidad de CO2 de la atmósfera. En la Figura 5 se proyectan en escalas logarítmicas las concentraciones de CO2 en la atmósfera y de CaCO3 en agua para distintas temperaturas. Se observa que, a una temperatura dada, a medida que aumenta la cantidad de CO2 aumenta la cantidad de CaCO3 disuelto, y que para una misma cantidad de CO2, la cantidad de CaCO3 disminuye con la temperatura. Esto mismo puede observarse en la Figura 8, donde la presión parcial de CO2 se proyecta respecto de la cantidad de CaCO3 disuelto a la temperatura de 16.1 oC.

Figura 7. Solubilidad de CO2 en el agua en función del CO2 disuelto en la atmósfera y la temperatura (tomado de Winkler, 1973).
El tratamiento preciso de los procesos de disolución de carbonatos incluye una serie de reacciones distintas de las anteriores (16) y (17), y es bastante complicado ya que entran muchas variables que modifican las cantidades disueltas de las distintas especies. Estas ecuaciones son las siguientes (los valores de logK son para 25 oC):
CaCO3 = Ca2+ + (CO3)= (logK = -8.3) (18)
H2CO3 = H+ + (HCO3)– (logK = -6.4) (19)
(HCO3)– = H+ + (CO3)= (logK = -10.3) (20)
H2O = H+ + (OH)– (logK = -14) (21)
CO2 + H2O= H2CO3 (logK = 6.48) (22)
Las variables involucradas son la temperatura, presión (en nuestro caso se puede asumir constante 1 atm), presión parcial de CO2, y la concentración de [H2CO3], [(HCO3)–], [(CO3)=], [H+], [(OH)–], y [Ca2+]. El tratamiento en equilibrio esta serie de ecuaciones químicas permite conocer las concentraciones de las distintas especies químicas en solución si fijamos el resto de las variables (P, T y PCO2). Por ejemplo, los cálculos a 25 oC indican que la concentración de Ca2+ unas tres veces mayor (10-3.4) en una solución en equilibrio con la atmósfera (presión parcial de CO2 atmosférico de 3.16*10-6 atm, correspondiente a una concentración de 0.034 g/l [1], y pH = 8.4), que en una solución sin aporte externo de CO2 ([CO2] = 0 g/l, pH = 10).
El tratamiento en equilibrio informará por lo tanto de las condiciones de saturación de las disoluciones, esto es, de las cantidades máximas de calcita que puede disolverse bajo unas condiciones dadas. Se pueden llegar a conocer las condiciones de pH ([H+]) y la cantidad de Ca2+ disuelto en equilibrio de las soluciones saturadas para distintas temperaturas. En base a estos cálculos se pueden predecir las condiciones de disolución y precipitación de los carbonatos. A mayor cantidad de CO2 disuelto, menor pH y menor temperatura, la solubilidad es mayor. Las curvas de Trombe (Figura 9) reflejan estas relaciones. Las aguas que se localizan en el campo subsaturado para las condiciones de pH y temperatura medidas se denominan "agresivas" (punto B del diagrama), dado que pueden disolver más cantidad de material. El resto de las agua se denominan saturadas (punto A). Estas curvas pueden dar una idea del grado de agresividad de una disolución determinada.
Para establecer relaciones cuantitativas precisas hay que asegurarse de las siguientes premisas:
· El sistema está en equilibrio.
· Deben conocerse las condiciones bajo las que se produjo la disolución.
· Las reacciones de disolución del carbonato son las anteriores.
· No existen otras reacciones independientes que puedan afectar al pH.
Estas constricciones afectan de distinta manera según la disolución proceda en la superficie de los materiales o en el interior del sistema poroso.
En el primer caso, es poco probable que el sistema esté en equilibrio, dado que las condiciones son muy cambiantes en pequeños intervalos de tiempo. Además, el agua superficial no suele estar en contacto con los materiales durante el tiempo suficiente para equilibrarse. En consecuencia, los cálculos anteriores son sólo indicativos de las cantidades máximas de carbonato que puede ser disuelto.

Figura 9. Curvas de Trombe a distintas temperaturas y con proyección de algunas aguas naturales.
En el segundo caso, si el agua contenida en el sistema poroso está suficiente tiempo en contacto con el material, es probable que se consiga el equilibrio. Sin embargo, las condiciones bajo las que tiene lugar la disolución suelen ser mal conocidas, dado que no existe acceso directo a las disoluciones contenidas dentro del sistema poroso. Si además existe conexión con el exterior, tanto atmosférico como con la fuente subterránea, es probable que existan gradientes de temperatura y/o de presión parcial de CO2, por lo que las tasas de disolución serán variables según los puntos del sistema poroso. Si existen además mezclas con aguas externas de composición distinta puede producirse lo que se denomina corrosión por mezcla de agua. Este fenómeno deriva de que las relaciones entre las cantidades de CaCO3 y CO2 disueltos en equilibrio el agua no guardan una relación lineal, sino exponencial. Estas relaciones se aprecian en la Figura 10. Así, si dos tipos de agua se mezclan se puede producir corrosión, aunque ambas estuviesen originalmente en equilibrio. Supongamos dos soluciones saturadas representadas por los puntos A y B. Cuando se mezclan, la composición resultante estará en algún lugar de la recta AB, en función de las proporciones relativas de la mezcla. Supongamos que la mezcla se proyecta en C. Este água no está en equilibrio, y por lo tanto debe liberar CO2 o disolver más CaCO3 para permanecer en equilibrio. De esta manera, la disolución del carbonato es posible, aún si no existe aporte externo de CO2. Las muestras L y M son sobresaturadas respecto al Ca2+, por lo que no producirán corrosión por mezcla, aunque bajo algunas circunstancias, como las que representan los puntos X e Y si puede producirse. La presencia de aguas sobresaturadas en CaCO3 respecto del CO2 es relativamente común, ya que la difusión del CO2 fuera de la disolución para reequilibrarse con otras condiciones es mucho más rápida que el depósito de calcita.
En resumen, la historia completa del agua en contacto con el material pétreo generalmente no podrá ser conocida lo suficientemente bien como para aplicar el tratamiento en equilibrio de manera precisa.

Figura 10. Representacción de la corrosión por mezcla de agua.
Otro aspecto que influye notablemente en la disolución de los carbonatos es el efecto de la actividad orgánica. Es muy posible que ciertas reacciones de quelación sean operativas bajo condiciones variadas, de forma que las tasas de disolución reales pueden ser contradictorias con las predichas por el tratamiento en equilibrio si se comparan distintos ambientes.
En cualquier caso, los procesos de disolución son el mecanismo de ataque químico más importante en carbonatos. Su efecto se deja notar más intensamente en las áreas polucionadas donde el contenido de CO2 es mayor y en zonas húmedas y frías, donde la capacidad de disolución del CO2 es mayor. Las formas de deterioro más comunes son disoluciones preferentes a lo largo de todo tipo de discontinuidades de las rocas calizas (e.g., fracturas, estilolitos, contactos entre bloque, cornisas, etc). Además, en el interior del material se producen pérdidas de materia con la consiguiente pérdida de cohesión. Costras, recristalizaciones de cementos carbonatados, eflorescencias y subeflorescencias, son formas de deterioro relacionadas con la precipitación de los carbonatos. En este sentido, las costras y eflorescencias, formadas a partir de soluciones saturadas o sobresaturadas provenientes del interior de la roca por reequilibración con el ambiente atmosférico externo y evaporación, pueden no redisolverse en el ambiente externo debido a los problemas antes mencionados de ausencia de equilibrio, aún cuando el ambiente externo fuese más agresivo.
Independientemente de los controles medioambientales, existe otro tipo de control propio del material, y es el referido a la composición del mismo. Por ejemplo, la presencia pequeñas cantidades de Mg en la calcita (menos del 1% respecto del contenido en Ca) incrementa el pH de una solución en equilibro en cerca de 0.1 unidades, por lo que la capacidad de disolución aumenta para una cantidad fija de CO2. Sin embargo, cuando el contenido de Mg aumenta más, ocurre lo contrario, de manera que la capacidad de disolución disminuye. Si en vez de calcita, el carbonato es dolomita, la capacidad de disolución es mucho más baja (Tabla).
Los carbonatos pueden contener algunos metales pesados en concentraciones muy bajas (se denominan trazas, por debajo de 0.1 - 0.01 % en peso) que tienen un importante efecto en la solubilidad. Los estudios al respecto indican que la presencia de estos elementos tiene un efecto inhibidor de la disolución. Esto puede observase en la Tabla 2, donde las concentraciones de Ca disuelto descienden a medida que aumenta el contenido de escandio (Sc), uno de los metales con mayor poder de inhibición conocidos. De hecho, la calcita parece que no continúa disolviéndose aún a pesar de que la solución no llega a la saturación para las condiciones experimentales (1 atm PCO2). Así, la solubilidad de la calcita parece reducirse a la mitad con sólo un 0.2 % de Sc respecto del Ca en la calcita. Los metales que actuan de esta manera son los siguientes, en orden decreciente de efectividad: Pb, La, Y, Sc, Cd, Cu, Au, Zn, Ge, Mn. Algunos, como el Pb, Sc y Cd, son comunes en los carbonatos.
Tabla 2. Solubilidad de la calcita en función del contenido en Sc.
Solubilidad (g mol/l) 9.0·10-3 8.6·10-3 7.2·10-3 4.8·10-3
Concentración de Sc (g mol/l) 0 10-7 10-6 10-5
No hay comentarios:
Publicar un comentario