CORRELACION ENTRE EL ASPIRADO MEDULAR, LA BIOPSIA ÓSEA Y EL INMUNOFENOTIPO EN LINFOMAS DE CÉLULA PEQUEÑA.
Dra Alicia Domingo-Claros / Dra. Esther Alonso / Dr. Vicente Romagosa*. Servicio de Hematología y *Anatomía Patológica. Hospital Universitari Prínceps d´Espanya. L´Hospitalet de Llobregat. Barcelona.
CASO 1
HISTORIA CLÍNICA:
Paciente de 46 años que se deriva a nuestro servicio para estudio de banda monoclonal. En los antecedentes destaca hace 5 años el diagnóstico de esofagitis grado I que se estudia a raíz de detectarse anemia discreta y aumento de la VSG (95mm/h). En una analítica de control (julio-00) se detecta VSG 143 mm/h, anemia: 99 g/L y VCM: 71.5 fL; en el proteinograma aparece componente homogéneo en la zona de las gammaglobulinas. El paciente relata astenia marcada.
Exploración física: anodina.
Pruebas complementarias: Hemograma: hemoglobina: 98g/L, Leucocitos: 4,2 x 109/L con recuento diferencial normal. Plaquetas: 266 x109/L. VSG: 120 mm/h.
En la bioquímica destaca: LDH: 4.9 mkat/L, b2-microglobulina: 5,6 mmol/L, proteínas totales: 62,5 g/L, componente monoclonal: 30g/L. Dosificación de inmunoglobulinas: IgG: 610 mg/dL, IgA: 23 mg/dL, IgM: 3.590 mg/dL. La inmunofijación en suero muestra cadenas pesadas mu y ligeras kappa. En orina se detecta proteinuria con presencia de banda monoclonal, y en la inmunofijación cadenas kappa monoclonales. Crioglobulinas negativas. VHB y VHC negativos.
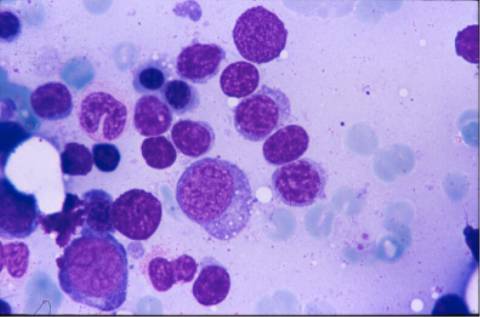
RX: La tomografía axial computerizada muestra adenopatías axilares bilaterales, mediastínicas y retroperitoneales. El estudio morfológico y fenotípico de los linfocitos en sangre periférica es normal. El aspirado medular muestra: una infiltración intersticial (50%), que tiende a agruparse en alguna zona, constituida por linfocitos de cromatina madura y citoplasma visible, mezclados con elementos de núcleo excéntrico y diferenciación plasmacítica; acompañamiento por células plasmáticas y mastocitos. La serie eritroide está conservada (29%) y la serie granulopoyética disminuida (17%); plasmáticas:4% y megacariocitos normales.
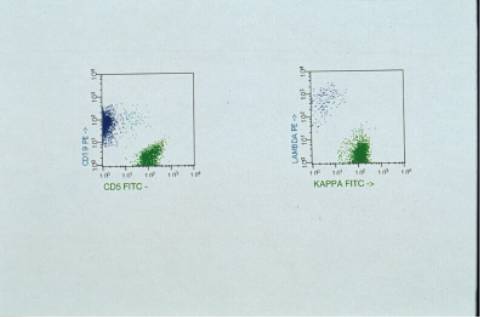
El inmunofenotipo en MO: la población linfoide global muestra un predominio de linfocitos T (35%) sobre B (19%); se aíslan las células CD19 que muestran CD5: neg, CD23: 15% (d), FMC-7: 80% (m), CD22: 95% (d), CD79b: 93% (m), CD38: 84% (m), CD20: 100% (m/i), CD11c: neg, CD25: neg, Kappa: 90% (m), lambda negativa; IgM negativa, IgD positiva. Una pequeña población coexpresa CD19/CD10. La biopsia ósea muestra una infiltración parcheada que tiende a confluir por linfocitos pequeños con diferenciación linfoplasmocitoide y células plasmáticas compatible con linfoma de linfocito de célula pequeña sugestivo de enfermedad de Waldeström.
ICONOGRAFÍA
|
|
 5 5: el inmunofenotipo realizado por citometría de flujo en la médula ósea al diagnóstico muestra un predominio de linfocitos T (35%) sobre linfocitos B (19%). Aislando la población B muestra positividad media para otros marcadores B como CD22, CD79b, CD20 y FMC-7, así como para CD38; negatividad para CD5, CD23 y CD10; restricción de IgS kappa de intensidad media, IgM negativa, IgD positiva.
|
|
|
 9 9: el inmunofenotipo de reevaluación que se realizó tras dos ciclos de 2-CDA muestra un predominio de linfocitos B (26%) sobre linfocitos T (22%) que son CD5 negativos y presentan un porcentaje muy superior de cadenas kappa sobre lambda, como al diagnóstico, lo que demuestra persistencia de población monoclonal.
|
Evolución:
Con el diagnóstico de enfermedad de Waldeström se inicia tratamiento por existir anemia importante de proceso crónico. Entra en un protocolo de 2-clordeoxiadenosina (2 ciclos) obteniéndose buena respuesta clínica con desaparición de la anemia y disminución del 40% del componente monoclonal. En la bioquímica se observa disminución de b2-microglobulina: 3,3 mg/L y de IgM: 1250 mg/dL.
Se realiza un aspirado de control que muestra escasa celularidad, sin grumo, infiltrado por linfocitos atípicos. El inmunofenotipo muestra un predominio de linfocitos B (26%) sobre los T ( 22%); aislando la población CD19 se detecta un predominio de cadenas kappa sobre lambda (34/5).
En el último control, disminución de la b2-microglobulina: 2,7 mg/L (normal < 2,4), disminución de la IgM: 910 mg/dL, LDH normales, Hb: 128 g/L, VCM: 83 fL, leucocitos: 3,7 x109/L con recuento normal, plaquetas: 144.0 x109/L. El AM no muestra infiltración por linfocitos atípicos y el inmunofenotipo muestra predominio de linfocitos T (53%) sobre B (23%) siendo la población B policlonal.
DISCUSIÓN:
El inmunocitoma o linfoma linfoplasmacítico de las clasificaciones REAL/OMS es un linfoma de célula pequeña. El diagnóstico diferencial debe hacerse con la LLC-B, el linfoma del manto, el linfoma folicular y los linfomas de la zona marginal. No siempre la morfología y el fenotipo son característicos por lo que muchas veces es un diagnóstico de exclusión. Cuando cursa con componente monoclonal IgM corresponde a la enfermedad de Waldeström, a la que se asocia clínica de hiperviscosidad por la gammapatía monoclonal, y en ocasiones fenómenos autoinmunes. Es un linfoma de curso indolente que puede transformarse a linfoma de célula grande inmunoblástico. En el aspirado medular nos encontramos con una infiltración linfoide intersticial que tiende a formar agregados, donde predominan las células pequeñas maduras mezcladas con algunas con diferenciación plasmacítica, algún elemento mas blástico y acompañamiento por células plasmáticas; la presencia de mastocitos que se consideraba en la tríada clásica celular del Waldeström los podemos ver en cualquier SLPC-B de célula pequeña. El inmunofenotipo se distingue de la LLC por ser CD5, CD10 neg CD23 variable y expresión intensa de marcadores B, de CD38 y de IgS. Puede expresar bcl-2 sin la t(14;18) y la expresión de KI-67 es baja lo que condiciona su curso clínico. El 50% cursan con la traslocación t(9;14)(p13;q32) que corresponde a la región que contiene el gen PAX-5. La biopsia ósea está infiltrada formando agregados linfoides mal delimitados e infiltración intersticial que algunos casos puede llegar a ser difusa; se suelen ver linfocitos maduros entremezclados con algunas células plasmáticas. La sangre periférica cursa con leucopenia y sin linfocitosis monoclonal aunque hay casos que cursan con expresión periférica simulando una LLC. Suele acompañarse de anemia y plaquetopenia que en algunas ocasiones es el motivo del estudio. No siempre cursan con adenopatías por lo que el estudio medular es lo que permitirá el diagnóstico.
En la mayoría de estos linfomas existe una buena correlación entre el aspirado medular, la biopsia ósea y el inmunofenotipo. Sin embargo, existen algunas situaciones como cuando se inicia la búsqueda de un síndrome linfoproliferativo en pacientes que presentan banda monoclonal IgM, o bien son VHC positivos o presentan fenómenos autoinmunes, en que podemos hallar agregados linfoides en médula ósea constituidos por linfocitos pequeños, maduros y con pocos elementos en transición hacia célula plasmática, es decir con una citología anodina que pueden simular un SLPC y en realidad tratarse de agregados linfoides benignos reactivos. Son enfermos que no presentan adenopatías por lo que no disponemos de histología ganglionar. En estos casos el estudio conjunto es imprescindible y será el inmunofenotipo que nos permitirá demostrar si existe una población B monoclonal. Otra peculiaridad de este linfoma de célula pequeña es que la infiltración por linfocitos B puede ser moderada y estar acompañada por una proporción importante de linfocitos T lo que por morfología, tanto en el aspirado como en la biopsia ósea, es difícil de detectar, y será el estudio inmunofenotípico que nos permitirá aislar la población B y demostrar la clonalidad mediante la restricción de IgS.pan en la inmunidad innata, son capaces de reconocer lo "propio" también tienen propiedades líticas.











No hay comentarios:
Publicar un comentario