Física molecular
El principio de Franck–Condon es una regla en espectroscopia y química cuántica que explica la intensidad de lastransiciones vibrónicas. Las transiciones vibrónicas son los cambios simultáneos en los niveles de energía electrónicos y vibracionales de una molécula debidos a la absorción o emisión de un fotón de la energía apropiada. El principio dicta que durante una transición electrónica, un cambio desde un nivel energético vibracional a otro será más probable que ocurra si las dos funciones de onda vibracionales se traslapan de manera significativa.
Información general


El principio de Franck-Condon tiene una bien establecida interpretación semi-clásica basada en las contribuciones originales de James Franck [Franck 1926]. Las transiciones electrónicas son esencialmente instantáneas comparadas con la escala de tiempo de los movimientos nucleares, por tanto si la molécula se va a mover a un nuevo nivel vibracional durante la transición electrónica, este nuevo nivel vibracional debe ser instantáneamente compatible con las posiciones nucleares y el momentum (cantidad de movimiento) del nivel vibracional de la molécula en el estado electrónico que lo origina. En la imagen semi-clásica de las vibraciones (oscilaciones) de un oscilador armónico, las condiciones necesarias pueden ocurrir en los puntos de retorno, donde el momentum es cero.
|
En la imagen mecánico cuántica, los niveles vibracionales y las funciones de onda vibracionales son las deloscilador armónico cuántico, o de aproximaciones más complejas de la energía potencial de las moléculas, tales como el potencial de Morse. La Figura 1 ilustra el principio de Franck–Condon para transiciones vibrónicas en una molécula con funciones de energía potencial tipo Morse tanto en los estados electrónicos basal y excitado. En la aproximación de baja temperatura, la molécula inicia en el nivel vibracional v = 0 del estado electrónico basal y al absorber un fotón de la energía necesaria, realiza una transición al estado electrónico excitado. La configuración electrónica del nuevo estado puede resultar en un desplazamiento de la posición de equilibrio de los núcleos que constituyen la molécula. En la figura, este desplazamiento en las coordenadas nucleares entre el estado basal y el primer estado excitado es etiquetado como q 01. En el caso más simple de una molécula diatómica el eje de las coordenadas nucleares se refiere a la separación internuclear. La transición vibrónica es indicada por una flecha vertical debido a la asunción de coordenadas nucleares constantes durante la transición. La probabilidad de que la molécula pueda terminar en cualquier nivel vibracional particular es proporcional al cuadrado del traslape (vertical) de las funciones de onda vibracionales de los estados original y final (véase debajo la sección de la formulación mecánico cuántica). En el estado electrónico excitado las moléculas rápidamente se relajan al más bajo nivel vibracional (regla de Kasha), y desde ahí pueden decaer al más bajo estado electrónico mediante la emisión de un fotón. El principio de Franck–Condon es aplicado igualmente a la absorción y a la fluorescencia.
La aplicabilidad del principio de Franck–Condon en la absorción y fluorescencia, junto con la regla de Kasha lleva a una aproximada simetría de espejo mostrada en la Figura 2. La estructura vibracional de las moléculas en un gas frío y escaso es más claramente visible debido a la ausencia de ensanchamiento no homogéneo de las transiciones individuales. Las transiciones vibrónicas son dibujadas en la Figura 2 como líneas Lorentzianas estrechas e igualmente espaciadas. El espaciado igual entre los niveles vibracionales es únicamente el caso para el potencialparabólico de osciladores armónicos simples; en potenciales más realistas, tales como los que se muestran en la Figura 1, el espaciamiento de la energía decrece con el incremento de la energía vibracional. Las transiciones electrónicas hacia y desde los estados vibracionales más bajos son ocasionalmente referidos como transiciones 0-0 (cero-cero) y tienen la misma energía tanto en la absorción como en la fluorescencia.
Desarrollo del principio
En un reporte publicado en 1926 por el Transactions de la Faraday Society, James Franck se interesó por los mecanismos de las reacciones químicas fotoinducidas. El presunto mecanismo era la excitación de una molécula por un fotón seguida de una colisión con otra molécula durante el corto periodo de excitación. La pregunta fue que si era posible para una molécula romperse en en fotoproductos en una sola etapa, la absorción de un fotón, y sin una colisión. Con el fin de que una molécula se rompa debe adquirir energía vibracional del fotón que exceda la energía de disociación, esto es, la energía para romper un enlace químico. Sin embargo, como era conocido en ese tiempo, las moléculas únicamente absorberán la energía correspondiente a las transiciones cuánticas permitidas y no hay niveles vibracionales arriba del nivel de energía de disociación del pozo de potencial. La absorción del fotón de alta energía lleva a una transición hacia un estado electrónico más alto en vez de a la disociación. Al examinar cuánta energía vibracional puede adquirir una molécula cuando es excitada a un nivel electrónico más alto, y si esta energía vibracional puede ser suficiente para romper inmediatamente la molécula, él dibujó tres diagramas representando los posibles cambios en la energía enlazante entre el estado electrónico más bajo y los estados electrónicos más altos.
|
James Franck reconoció que los cambios en los niveles vibracionales pudieran ser consecuencia de la naturaleza instantánea de la excitación hacia niveles de energía electrónica más altos y una nueva posición de equilibrio para el potencial de interacción nuclear. Edward Condon extendió esta observación más allá de las fotorreacciones en un artículo en el Physical Review de 1926 titulado "A Theory of Intensity Distribution in Band Systems" (una teoría de la distribución de intensidad en sistemas de bandas). Aquí él realiza la formulación semi-clásica de una manera bastante similar a la forma moderna. La primer referencia conjunta tanto a Franck como a Condon en cuanto al nuevo principio apareció en la misma publicación de 1926 del Physical Review en un artículo sobre la estructura de bandas del monóxido de carbono por Raymond Birge.

Formulación mecánico cuántica
Considérese una transición dipolar eléctrica desde el estado vibracional inicial (υ) del nivel electrónico basal (ε),  , hacia algún estado vibracional (υ') de un estado electrónico excitado (ε'),
, hacia algún estado vibracional (υ') de un estado electrónico excitado (ε'),  (véase notación Bra-Ket). El operador de dipolo molecular μ es determinado por la carga (-e) y las localizaciones (ri) de los electrones así como las cargas (+eZj) y las localizaciones (Rj) de los núcleos:
(véase notación Bra-Ket). El operador de dipolo molecular μ es determinado por la carga (-e) y las localizaciones (ri) de los electrones así como las cargas (+eZj) y las localizaciones (Rj) de los núcleos:
, hacia algún estado vibracional (υ') de un estado electrónico excitado (ε'), (véase notación Bra-Ket). El operador de dipolo molecular μ es determinado por la carga (-e) y las localizaciones (ri) de los electrones así como las cargas (+eZj) y las localizaciones (Rj) de los núcleos: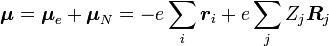
La amplitud de probabilidad P para la transición entre estos dos estados está dada por

Donde  y
y  son, respectivamente, las funciones de onda globales del estado inicial y final. Las funciones de onda globales son el producto de las funciones de onda vibracional (dependiente de las coordenadas espaciales de los núcleos) y electrónicas espaciales y de espín individuales:
son, respectivamente, las funciones de onda globales del estado inicial y final. Las funciones de onda globales son el producto de las funciones de onda vibracional (dependiente de las coordenadas espaciales de los núcleos) y electrónicas espaciales y de espín individuales:
y son, respectivamente, las funciones de onda globales del estado inicial y final. Las funciones de onda globales son el producto de las funciones de onda vibracional (dependiente de las coordenadas espaciales de los núcleos) y electrónicas espaciales y de espín individuales: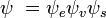
Esta separación de las funciones de onda electrónicas y vibracionales es una expresión de la aproximación de Born-Oppenheimer y es la asunción fundamental del principio de Franck–Condon. Combinando estas ecuaciones se llega a una expresión para la amplitud de probabilidad en términos de contribuciones separadas electrónicas, de espín y vibracionales:




La parte independiente de espín de la integral inicial está aquí aproximada como un producto de dos integrales:

Esta factorización sería exacta si la integral 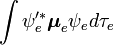 sobre las coordenadas espaciales de los electrones no dependiera de las coordenadas nucleares. Sin embargo, en la aproximación de Born-Oppenheimer
sobre las coordenadas espaciales de los electrones no dependiera de las coordenadas nucleares. Sin embargo, en la aproximación de Born-Oppenheimer  y
y 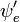 dependen (paramétricamente) de las coordenadas nucleares, así que la integral (llamada superficie de transición dipolar) es una función de las coordenadas nucleares. Debido a que la dependencia es usualmente más algo pequeña se omite (es decir, la asunción de que la superficie de transición dipolar es independiente de las coordenadas nucleares, llamada la aproximación de Condon) es a veces permitida.
dependen (paramétricamente) de las coordenadas nucleares, así que la integral (llamada superficie de transición dipolar) es una función de las coordenadas nucleares. Debido a que la dependencia es usualmente más algo pequeña se omite (es decir, la asunción de que la superficie de transición dipolar es independiente de las coordenadas nucleares, llamada la aproximación de Condon) es a veces permitida.
sobre las coordenadas espaciales de los electrones no dependiera de las coordenadas nucleares. Sin embargo, en la aproximación de Born-Oppenheimer y dependen (paramétricamente) de las coordenadas nucleares, así que la integral (llamada superficie de transición dipolar) es una función de las coordenadas nucleares. Debido a que la dependencia es usualmente más algo pequeña se omite (es decir, la asunción de que la superficie de transición dipolar es independiente de las coordenadas nucleares, llamada la aproximación de Condon) es a veces permitida.
La primera integral después del signo de más es igual a cero debido a que las funciones de onda electrónicas de estados diferentes son ortogonales. Lo que queda es el producto de tres integrales. La primera integral es la integral de traslape vibracional, también llamada el factor de Franck-Condon. Las dos integrales restantes que contribuyen a la amplitud de probabilidad determinan las reglas de selección electrónica espacial y de espín.
El principio de Franck–Condon es un enunciado sobre las transiciones vibracionales permitidas entre dos estados electrónicos diferentes, otras reglas de selecciónmecánico cuánticas puedes disminuir la probabilidad de una transición o prohibirla definitivamente. Las reglas de selección rotacionales han sido omitidas en la derivación superior. Las contribuciones rotacionales pueden ser observadas en los espectros de gases pero son fuertemente suprimidas en líquidos y sólidos.
Debería ser claro que la formulación mecánico cuántica del principio de Franck–Condon es el resultado de una serie de aproximaciones, principalmente la asunción de la transición dipolar eléctrica y la aproximación de Born-Oppenheimer. Las más débiles transiciones electrónicas de dipolo magnético y cuadrupolo eléctrico junto con la incompleta validez de la factorización de la función de onda total en funciones de onda nuclear, electrónica espacial y de espín significa que las reglas de selección, incluyendo el facto de Franck-Condon, no son estrictamente observadas. Para cualquier transición dada, el valor de P es determinado por todas las reglas de selección, sin embargo la selección de espín es el mayor contribuyente, seguido por las reglas de selección electrónicas. El factor de Franck-Condon solo modula débilmente la intensidad de las transiciones, es decir, contribuye con un factor del orden de 1 a la intensidad de las bandas cuyo orden de magnitud es determinado por las otras reglas de selección. La tabla debajo da el rango de los coeficientes de extinción para las posibles combinaciones de las reglas de selección de espín y orbital permitidas y prohibidas.
| Rango de los coeficientes de extinción (ε) (mol−1 cm−1) | |
|---|---|
| Permitidas por espín y orbitalmente | 103 a 105 |
| Permitidas por espín pero orbitalmente prohibidas | 100 a 103 |
| Prohibidas por espín pero orbitalmente permitidas | 10−5 a 100 |
Metáforas de Franck-Condon en espectroscopia
El principio de Franck–Condon, en su forma canónica, se aplica únicamente a cambios en los niveles vibracionales de una molécula en el curso de un cambio en los niveles electrónicos ya sea mediante la absorción o la emisión de un fotón. La intuición física de este principio está anclada por la idea de que las coordenadas nucleares de los átomos que constituyen la molécula no tienen tiempo de cambiar durante la muy breve cantidad de tiempo involucrado en una transición electrónica. Sin embargo, esta intuición física puede ser, y de hecho lo es, rutinariamente extendida a las interacciones entre las moléculas absorbentes o emisoras de luz (cromóforos) y sus ambientes. Las metáforas de Franck-Condon son apropiadas porque las moléculas ocasionalmente interactúan fuertemente con las moléculas de los alrededores, particularmente en líquidos y sólidos, y estas interacciones modifican las coordenadas nucleares del cromóforo en formas que son cercanamente análogas a las vibraciones moleculares consideradas por el principio de Franck–Condon.

Principio de Franck-Condon para fonones
La analogía más cercana de Franck-Condon es debida a la interacción de fonones (cuantos de vibraciones de red) con las transiciones electrónicas de cromóforos embebidos como impurezas de la red. En esta situación, las transiciones a niveles electrónicos más altos pueden tener lugar cuando la energía del fotón corresponde a la energía de transición puramente electrónica o la energía de transición puramente electrónica más la energía de uno o más fonones de la red. En la aproximación de baja temperatura, la emisión viene del nivel fonón-cero del estado excitado hasta el nivel fonón-cero del estado basal o niveles fonónicos más altos que el estado basal. Así como en el principio de Franck–Condon, la probabilidad de las transiciones que involucran fonones es determinada por el traslape de las funciones de onda de fonón en los niveles de energía inicial y final. Para el principio de Franck–Condon aplicado a transiciones de fonón, la etiqueta del eje horizontal de la Figura 1 es reemplazada en la Figura 6 con la coordenada configuracional para un modo normal. La energía potencial del modo de red  en la Figura 6 es representada como la de un oscilador armónico, y el espaciado entre niveles de fonón (
en la Figura 6 es representada como la de un oscilador armónico, y el espaciado entre niveles de fonón ( ) es determinado por los parámetros de red. Debido a que la energía de los fonones individuales es generalmente bastante pequeña, las transiciones de fonón-cero o menores únicamente puedes ser observadas a temperaturas debajo de alrededor de 40 kelvin.
) es determinado por los parámetros de red. Debido a que la energía de los fonones individuales es generalmente bastante pequeña, las transiciones de fonón-cero o menores únicamente puedes ser observadas a temperaturas debajo de alrededor de 40 kelvin.
en la Figura 6 es representada como la de un oscilador armónico, y el espaciado entre niveles de fonón () es determinado por los parámetros de red. Debido a que la energía de los fonones individuales es generalmente bastante pequeña, las transiciones de fonón-cero o menores únicamente puedes ser observadas a temperaturas debajo de alrededor de 40 kelvin.Principio de Franck-Condon en solvatación

Las consideraciones de Franck-Condon pueden también ser aplicadas a las transiciones electrónicas de los cromóforos disueltos en líquidos. En este uso de la metáfora de Franck-Condon, los niveles vibracionales de los cromóforos, así como las interacciones de los cromóforos con fonones en el líquido, contribuyen a la estructura de los espectros de absorción y emisión, pero estos efectos son considerados de manera separada e independiente.
Considérese los cromóforos rodeados por moléculas de disolvente. Estas moléculas circundantes pueden interactuar con los cromóforos, particularmente si las moléculas del disolvente son polares. Esta asociación entre disolvente ysoluto es referida como solvatación y es una interacción estabilizadora, esto es, las moléculas de disolvente pueden moverse y rotar hasta que la energía de la interacción sea minimizada. La interacción en sí misma involucra fuerzaselectrostáticas y fuerzas de Van der Waals y puede incluir también enlaces de hidrógeno. Los principios de Franck–Condon pueden ser aplicados cuando las interacciones entre los cromóforos y las moléculas circundantes de disolvente sean diferentes en los estados electrónicos basal y excitado. Este cambio en la interacción se puede originar, por ejemplo, debido a momentos dipolares diferentes en estos dos estados. Si el cromóforo comienza en su estado basal y se acerca al equilibrio con las moléculas de disolvente que lo rodean, y entonces absorbe un fotón que lo lleva al estado excitado, su interacción con el disolvente estará lejos del equilibrio en el estado excitado. Este efecto es análogo al principio de Franck–Condon original: la transición electrónica es muy rápida comparada con el movimiento de los núcleos —el rearreglo de las moléculas de disolvente en el caso de la solvatación—. Ahora se habla de una transición vertical, pero ahora la coordenada horizontal es el espacio de interacción disolvente-soluto. Este eje coordenado es a veces llamado "Coordenada de Solvatación" y representa, de manera un poco abstracta, todas las dimensiones de movimiento relevantes de todas las moléculas de disolvente interactuantes.
En el principio de Franck–Condon original, después de la transición electrónica, las moléculas que terminan en estados vibracionales superiores inmediatamente comienzan a relajarse al menor estado vibracional. En el caso de la solvatación, las moléculas de disolvente inmediatamente intentarán arreglarse ellas mismas para minimizar la energía de interacción. La tasa de relajación del disolvente depende de su viscosidad. Asumiendo que el tiempo de la relajación del disolvente es corto comparado con el tiempo de vida del estado excitado electrónico, la emisión será desde el estado energético más bajo del disolvente del estado electrónico excitado. Para disolventes de moléculas pequeñas tales como el agua o el metanol a temperatura ambiente, el tiempo de relajación del solvente es del orden de algunas decenas de picosegundo mientras que los tiempos de vida del estado excitado del cromóforo varían desde unos pocos picosegundos hasta unos pocos nanosegundos. Inmediatamente después de la transición al estado electrónico basal, las moléculas de disolvente deben también rearreglarse para acomodar la nueva configuración electrónica del cromóforo. La Figura 7 ilustra el principio de Franck–Condon aplicado a la solvatación. Cuando la disolución es iluminada con luz correspondiente a energía de transición electrónica, algunos de los cromóforos se moverán al estado excitado. Dentro de este grupo de cromóforos habrá una distribución estadística de las energías de interacción disolvente-cromóforo, representadas en la figura por una función dedistribución normal. La interacción disolvente-cromóforo es dibujada como un potencial parabólico en ambos estados electrónicos. Debido a que la transición electrónica es esencialmente instantánea en la escala de tiempo del movimiento del disolvente (flecha vertical), la colección de cromóforos de estado excitado está inmediatamente lejos del equilibrio. El rearreglo de las moléculas de disolvente de acuerdo a la nueva curva de energía potencial es representado por las flechas curvadas en la Figura 7. Nótese que mientras las transiciones electrónicas están cuantizadas, la energía de interacción cromóforo-disolvente es tratada como un continuo clásico debido al gran número de moléculas involucradas. Aunque la emisión es representada como que toma lugar desde el mínimo del potencial de interacción cromóforo-disolvente del estado excitado, una emisión significativa puede tomar lugar antes de alcanzarse el equilibrio cuando la viscosidad del disolvente es alta o el tiempo de vida del estado excitado es corto. La diferencia de energía entre los fotones absorbidos y emitidos representada en la Figura 7 es la contribución de solvatación del desplazamiento de Stokes.

Figura 1. Diagrama de energía del principio de Franck–Condon. Debido a que las transiciones electrónicas son muy rápidas comparadas con los movimientos nucleares, los niveles vibracionales son favorecidos cuando corresponden a un cambio mínimo en las coordenadas nucleares. Los pozos de potencial son mostrados favoreciendo las transiciones entre v = 0 y v = 2.
No hay comentarios:
Publicar un comentario